The majority of eye medical conditions arise in adulthood rather than in childhood or infancy. When vision is significantly reduced during infancy or early childhood the most common causes include:
Certain ocular abnormalities (ex. congenital cataracts or high refractive errors) are easily recognizable at an initial eye examination. However many inherited retinal diseases such as Leber Congenital Amaurosis, Congenital Night Blindness or cortical vision impairment may require extensive eye testing to establish an accurate diagnosis. It is well known that in pediatric ophthalmology a routine ophthalmological evaluation may not be sufficient for establishing the underlying cause of visual disturbance. Electrophysiological exams of visual system, brain imaging, or even genetic testing may be warranted to confirm specific disorders or for cases involving neurodevelopmental disorders. We draw your attention to the fact that the majority of published information was taken from peer reviewed journals and books and trustworthy sources then adapted for the non-medical community because the team at Restore Vision Clinic in Berlin has taken a mission to educate their readers and potential patients.
We draw your attention to the fact that the majority of published information was taken from peer reviewed journals and books and trustworthy sources then adapted for the non-medical community because the team at Restore Vision Clinic in Berlin has taken a mission to educate their readers and potential patients.
Infancy or early childhood:
|
|
|
|
Preschool period:
|
Early grade school:
|

|
 An initial eye examination by eye care providers reveals clues about the underlying cause of the visual disturbance. Below we have collected some helpful signs which suggest potential ocular and/or visual abnormalities. Some of them are: congenital nystagmus, pupillary response to light, nearsightedness or farsightedness, photophobia (light sensitivity), “oculodigital sign”, overlooking, excess head and eye movements
An initial eye examination by eye care providers reveals clues about the underlying cause of the visual disturbance. Below we have collected some helpful signs which suggest potential ocular and/or visual abnormalities. Some of them are: congenital nystagmus, pupillary response to light, nearsightedness or farsightedness, photophobia (light sensitivity), “oculodigital sign”, overlooking, excess head and eye movements
For instance, (1) congenital nystagmus 𑁋 characterized by repetitive, uncontrolled shaking of the eyes 𑁋 is a common finding associated with eye or anterior (front) brain visual pathway disorders, but is rare in children with cortical visual impairment (CVI). Even when the eyes are structurally normal, children with nystagmus typically have an underlying visual sensory condition. Therefore if nystagmus is present in a child with visual impairment, CVI is an unlikely diagnosis but another ocular or neurological explanation likely exists.
Another simple, yet informative, diagnostic test able to be performed on infants is pupil evaluation. This entails observing the (2) pupillary response to light, one eye at a time. Pupil responses to light are usually brisk and large. While inherited retinal diseases often cause minimal, sluggish pupil responses, normal pupillary responses are generally observed in children with CVI. Furthermore, certain conditions, such as achromatopsia and congenital stationary night blindness (CSNB), may cause “paradoxical pupillary response” where the pupil constricts in darkness, rather than light.
Certain eye conditions are associated with unusually high refractive errors 𑁋 (3) nearsightedness or farsightedness. Leber congenital amaurosis (LCA) is usually accompanied by farsightedness while nearsightedness is a common feature of CSNB and other inherited retinal diseases. However, this data is not always useful as some conditions, such as ocular albinism, are linked with both farsightedness and nearsightedness.
Certain ocular conditions cause various degrees of (4) photophobia (light sensitivity) or aversion to light. Extreme avoidance of light is the hallmark of conditions like achromatopsia and ocular albinism. Significant photophobia is also witnessed in children with LCA and cone-rod or rod-cone dystrophies. Meanwhile optic nerve hypoplasia and autosomal dominant optic atrophy (ADOA) cause mild photophobia. Glare and photophobia is also observed in patients with media opacities, such as cataracts or corneal scars. Not exclusively due to ocular conditions though, photophobia may also be caused by neurological conditions such as migraines, head trauma, meningitis, brain hemorrhages, nerve damage, strokes, or brain tumors, and even CVI.
One hallmark of LCA is the (5) “oculodigital sign” described as poking, pressing, or rubbing the eyes with fingers or knuckles. This sign may also be observed in other retinal conditions (retinopathy of prematurity, retinal dysplasia, cone-rod dystrophy), but not in cases of media opacities, optic nerve disease, or CVI. Interestingly, the oculodigital sign generally only presents if both eyes are affected, rather than just one eye.
(6) Overlooking. If a child’s central vision is missing, known as having a central scotoma, then the child will not be able to look directly at objects, but rather, will have to look adjacent to the object in order to see it. This is sometimes referred to as overlooking and occurs in retinal conditions which affect the center area of the retina called the macula. Macular diseases affect central vision, but may leave mid-peripheral and peripheral vision intact.
(7) Excess head and eye movements. Objecting tracking and fixation patterns differ by extent of vision loss, which may be indicative of certain ocular or neurological conditions. Although somewhat dependent on age, children with 20/200 or better vision tend to track objects using eye movements while children with worse than 20/200 vision track objects using head and eye movements. Use of head movements for object tracking is more prevalent in cases of severe visual deficits. A child’s eye fixation (ability to focus their gaze on an object) may also be affected in cases of diminished eyesight. For children with severe vision loss, assessment of visual skills such as object tracking and fixation patterns will likely reveal expected deficiencies. Additional testing may be warranted to more accurately determine the status of the child’s overall visual function.
Although the visual assessment portion of an eye examination may both objectively and subjectively estimate visual function, this does not always accurately portray the extent of the visual impairment experienced in real world situations. Collaboration between ophthalmologists, optometrists, neurologists, psychologists, pediatricians, and various therapists may enhance understanding of overall visual impairment and reinforce supportive management. The developmental, educational, psychological, and social demands of the child should be continuously reassessed and supported in addition to the visual components.
Dilation may be used to obtain clear views of the optic nerve, retina, and blood vessels located in the back of the eye. Children with visual impairment may have optic nerves that appear pale or underdeveloped. The retina may also have an underdeveloped or abnormally pigmented appearance. Retinal blood vessels may look unusually thin or thick in nature. However, if the source of reduced vision is of neurological origin, rather than ocular, the back of the eye may appear normal.
The normal appearance of the optic disc is somewhat age-dependent. While normal optic discs in adults are typically slightly pink in nature, in young infants the optic discs may demonstrate a slight gray or pale appearance. A gray or pale optic nerve in an adult is generally a sign of optic atrophy, yet in a newborn this finding may be benign. For this reason, a diagnosis of optic atrophy in a young infant should not be based only on optic disc appearance, but also on other relevant clinical and visual findings.
Likewise, a normal visual acuity doesn’t necessarily exclude the possibility of optic atrophy in children as optic atrophy does not always result in a reduction of central visual acuity. Similar to adults, certain optic disc appearances may lead to a clinician mistakenly diagnosing optic atrophy. This may occur in cases of large optic disc parameters, physiological cupping of the optic disc, deep physiologic cups, or staphylomas from axial myopia.
Signs of true optic atrophy may be visible in the peripapillary retinal nerve fiber layer. This may be visible as peripapillary nerve fiber layer dropout, sectoral or wedge defects, and generalized thinning of the retinal nerve fiber layer. Additionally, selective dropout of the nasal nerve fiber layer is suggestive of band atrophy, signifying damage to the axons crossing at the optic chiasm. Retinal nerve fiber layer defects may be visible with dilated fundus examination but are generally more apparent with optical coherence tomography (OCT). Other ophthalmic findings which may be indicative of optic atrophy include attenuation of retinal vessels, altered foveal light reflex, or loss of Gunn’s dots above retinal vessels.
Generally optic disc appearance does not prove useful in localizing a specific damaged visual pathway site. An exception is band atrophy, demonstrating injury to axons which decussate at the optic chiasm and may suggest a congenital suprasellar tumor. Band atrophy presents as a horizontal band of pallor, also called a “bow-tie” pattern, extending across the optic disc nasally to temporally. Band atrophy may have associated nasal retinal nerve fiber layer dropout. Exclusively found in chiasmal injury, bilateral band atrophy will result in a bitemporal hemianopia (or loss of temporal vision in both eyes). This injury is commonly due to compression from a suprasellar tumor. Unilateral band atrophy less specifically typically signifies a retrogeniculate injury with transsynaptic degeneration although it may reflect a pregeniculate injury occasionally. Congenital band atrophy in children is often a result of congenital optic tract syndrome, arteriovenous malformation, porencephaly, or ganglioneuromas. Any acquired band atrophy of only one optic disc occurs due to new injury to the contralateral optic tract near the optic chiasm.
Congenital nystagmus is an involuntary oscillating movement of the eyes typically presenting around 3-4 months of age. The term congenital nystagmus is a misnomer as it is typically not present immediately after birth. These children do not experience oscillopsia (perception of the world shaking) as the brain is capable of adapting to these rapid eye movements if present at an early age. Congenital nystagmus may occur in children with poor or normal vision. Nystagmus should not be confused with “roving” or “drifting” eye movements characterized by slow drifting of the eyes back and forth. Roving occurs due to the inability to fixate one’s gaze and may be indicative of more severe vision loss. Nystagmus should also not be confused with strabismus which is an eye turn that may occur constantly or intermittently, inward or outward. Nystagmus is a rapid shaking of the eyes either horizontally or vertically.

Nystagmus may occur in different forms 𑁋 jerk, pendular, or both depending on the direction of gaze. Nystagmus generally occurs due to defective afferent visual pathway processes, that is signaling from the eyes to the brain.
... More relevant and ubiquitous symptoms amongst kids with CSNB include photophobia, nystagmus, nearsightedness, and reduced central and peripheral vision. Some children with CSNB may also have a strabismus (eye turn).
 Congenital stationary night blindness (CSNB) is a collection of non-progressive inherited retinal diseases (IRD) characterized by impaired night vision, reduced central and peripheral vision, nystagmus, nearsightedness (myopia), and strabismus (eye turn). CSNB may be classified by both retinal appearance and electrophysiology findings.
Congenital stationary night blindness (CSNB) is a collection of non-progressive inherited retinal diseases (IRD) characterized by impaired night vision, reduced central and peripheral vision, nystagmus, nearsightedness (myopia), and strabismus (eye turn). CSNB may be classified by both retinal appearance and electrophysiology findings.
The electrophysiologic waveforms elicited by flashing light stimuli can further delineate CSNB as “complete” or “incomplete” depending on the signaling deficiencies of the rod photoreceptors and the ON and OFF bipolar cells.
Initial diagnosis of CSNB is made through signs and symptoms, ophthalmological examination, electrophysiology (visual evoked potentials and electroretinography). Exact molecular diagnosis is made through genetic testing. Ophthalmological examination along with diagnostic testing differentiates CSNB from other ocular conditions on the differential diagnosis list 𑁋 ocular albinism, achromatopsia, blue-cone monochromacy, X-linked motor nystagmus, X-linked juvenile retinoschisis.
Despite the name congenital stationary night blindness, children with CSNB are not actually completely blind in dim-lit situations. Deficiencies in rod photoreceptors, which are responsible for dim-lit vision, do cause diminished vision in dark environments, but usually do not result in complete blindness. Modern day adjustable lighting is helpful in alleviating much of these dim-lighting visual concerns. Furthermore, the extent and severity of night blindness varies by CSNB subtype with some forms (CSNB2) having only a mild reduction in dim-lit vision. For this reason, some researchers and clinicians have proposed changing the name of Congenital Stationary Night Blindness as to not inaccurately describe the condition for many cases. More relevant and ubiquitous symptoms amongst kids with CSNB include photophobia, nystagmus, nearsightedness, and reduced central and peripheral vision. Some children with CSNB may also have a strabismus (eye turn). The clinical course and degree of visual impairment of CSNB is “stationary” and therefore does not worsen over time.
 CSNB has multiple inheritance patterns including autosomal dominant, autosomal recessive, and X-linked. Numerous pathological variants also exist for CSNB. Pathological variants are genetic alterations which predispose an individual to a particular disease or condition through altering normal protein function or protein interactions. Pathological variants for CSNB include CABP4, GNB3, GPR179, GRM6, LRIT3, SAG, SLC24A1, TRPM1, GNAT1, PDE6B, RHO, CACNA1F, and NYX. As with all inherited retinal diseases, genetic testing is beneficial in confirming the diagnosis, determining the specific gene(s) at play, ascertaining the carrier status of the child and family members, and deciding best treatment approaches.
CSNB has multiple inheritance patterns including autosomal dominant, autosomal recessive, and X-linked. Numerous pathological variants also exist for CSNB. Pathological variants are genetic alterations which predispose an individual to a particular disease or condition through altering normal protein function or protein interactions. Pathological variants for CSNB include CABP4, GNB3, GPR179, GRM6, LRIT3, SAG, SLC24A1, TRPM1, GNAT1, PDE6B, RHO, CACNA1F, and NYX. As with all inherited retinal diseases, genetic testing is beneficial in confirming the diagnosis, determining the specific gene(s) at play, ascertaining the carrier status of the child and family members, and deciding best treatment approaches.
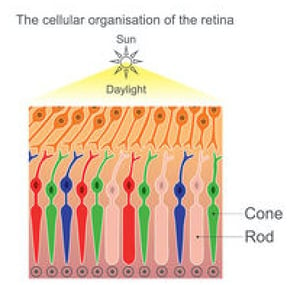 Achromatopsia, or rod monochromatism, is characterized by a functional absence of all three cone photoreceptor subtypes from birth. One major function of cone photoreceptors is color vision distinction, explaining why achromatopsia is sometimes referred to as total color blindness. Worldwide this condition affects approximately 1/30,000 persons.
Achromatopsia, or rod monochromatism, is characterized by a functional absence of all three cone photoreceptor subtypes from birth. One major function of cone photoreceptors is color vision distinction, explaining why achromatopsia is sometimes referred to as total color blindness. Worldwide this condition affects approximately 1/30,000 persons.
Symptoms of achromatopsia include poor central visual acuity and pendular nystagmus with the hallmark symptom being intense photophobia (light sensitivity). Though these children will display visual impairment, usually the most noticeable symptom will be extreme aversion to light. Visual acuity generally settles around 20/200 vision in optimal lighting, but is functionally worse in bright lighting due to the photophobia. The nystagmus may become less noticeable with age.
Currently, six different causative genetic variants have been connected with achromatopsia 𑁋 CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, and ATF6. Approximately 50% of cases are associated with CNGB3 mutations while 25% of cases are due to CNGA3 mutations. Each of these genes affect the retinal transduction pathway, specifically impairing cone photoreceptor hyperpolarization. Achromatopsia may also be acquired, occurring due to head trauma damaging the thalamus or cerebral cortex. Acquired achromatopsia is sometimes referred to as cerebral achromatopsia.
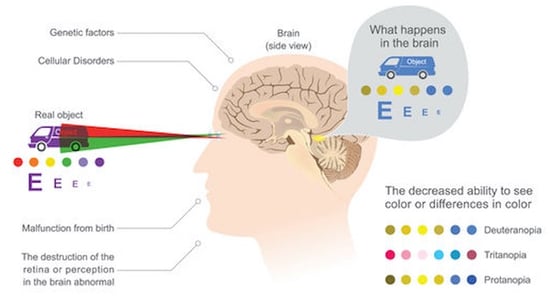 The deficient cone photoreceptor signaling in congenital achromatopsia is present at birth meaning the visual cortex never receives normal visual input. For this reason the neurological wiring, organization, and processing of the visual cortex never develops properly. The term for this abnormal input and processing leading to vision loss is amblyopia. Although best treated in young children, some evidence exists that amblyopia may be partially treatable in adults depending on the level of plasticity of the visual cortex, which is controversial. At least one study suggests that visual function may be improved in adult patients with achromatopsia. Treatment aims to improve both the cone photoreceptor signaling deficient in achromatopsia as well as the amblyopia caused by the achromatopsia.
The deficient cone photoreceptor signaling in congenital achromatopsia is present at birth meaning the visual cortex never receives normal visual input. For this reason the neurological wiring, organization, and processing of the visual cortex never develops properly. The term for this abnormal input and processing leading to vision loss is amblyopia. Although best treated in young children, some evidence exists that amblyopia may be partially treatable in adults depending on the level of plasticity of the visual cortex, which is controversial. At least one study suggests that visual function may be improved in adult patients with achromatopsia. Treatment aims to improve both the cone photoreceptor signaling deficient in achromatopsia as well as the amblyopia caused by the achromatopsia.
Achromatopsia typically follows an autosomal recessive inheritance pattern, meaning unaffected parents may be unknown carriers. The most causative genes are CNGA3 (20-30% of total cases) and CNGB3 (40-50% of total cases). Other pathological variants include CNGA3, CNGB3, GNAT2, PDE6C, ATF6 and PDE6H.
As with all genetic testing for IRDs there are four result possibilities:
Optical coherence tomography (OCT) is often utilized in the diagnosis and management of achromatopsia. At first glance, the OCT scan may appear fairly normal and inconsistent with the visual symptoms of the patient. This is due to achromatopsia affecting only a small physical portion of the retina and only one cell type (cone photoreceptors). The OCT results may even be normal 𑁋 demonstrating an intact IS/OS junction of the photorector layer and a normal outer nuclear layer. In more prominent cases or upon closer inspection of the OCT scan, one may see a broad and shallow foveal depression (possibly from hypoplasia or maldevelopment), mild foveal disruption, loss of the IS/OS junction, RPE attenuation in the macula, or a hyporeflective and optically void space in the photoreceptor layer corresponding to photoreceptor layer disruption.
 The Latin root word of albinism is “albus”, meaning white. Albinism is a congenital disorder characterized by decreases in melanin of not only the skin and hair, but also the pigmentation in the eye. Albinism of the eye only is known as ocular albinism (OA) while albinism of the eyes, skin, and hair is referred to as oculocutaneous albinism (OCA). Albinism occurs due to deficiencies in proteins or enzymes which contribute to melanin synthesis lead to markedly reduced or even absent pigmentation.
The Latin root word of albinism is “albus”, meaning white. Albinism is a congenital disorder characterized by decreases in melanin of not only the skin and hair, but also the pigmentation in the eye. Albinism of the eye only is known as ocular albinism (OA) while albinism of the eyes, skin, and hair is referred to as oculocutaneous albinism (OCA). Albinism occurs due to deficiencies in proteins or enzymes which contribute to melanin synthesis lead to markedly reduced or even absent pigmentation.
The worldwide incidence of albinism is approximately 1:5000 persons, however the incidence is as high as 1:1000 persons in areas of Africa and as low as 1:37000 in the United States suggesting genetic influence.
Ocular albinism (OA) presents as hypopigmentation of the pigmented areas of the eye 𑁋 iris, retinal pigment epithelium (RPE), and choroid. OA is a permanent, although non-progressive, disorder of ocular hypopigmentation in conjunction with visual pathway abnormalities. Amblyopia, due to input and processing anomalies of the optic nerves and visual pathways, contributes to the reduced vision experienced by these patients. Symptoms of OA include poor central and peripheral vision, extreme photophobia (light sensitivity), strabismus (eye turn), nystagmus (involuntary oscillating eye movements), and reduced binocularity (eye teaming). Refractive errors are common but may be myopic, hyperopic, or astigmatic. Visual acuity is highly variable ranging from minimally affected to legally blind and is usually associated with the degree of hypopigmentation.
One hallmark of OA is transillumination defects seen through slit lamp retroillumination which are indicative of thin or absent iris pigmentation. With dilated fundus examination, a “blond fundus” is observable due to hypopigmentation of the underlying choroid. Optical coherence tomography (OCT) demonstrates foveal hypoplasia or underdevelopment of the central retina. Results of visual evoked potentials (VEP), a type of electrophysiology, may be abnormal due to optic nerve miswiring. The eyelashes or eyebrows may be white or blond in color also.
 Interestingly, melanin and pigmented cells appear to play a role in the development of the visual pathway from the eye to the brain, specifically at the optic nerve and chiasmal decussation behind the optic nerve. The reduction or absence of this pigment is believed to result in a misrouting of optic nerve axons at the optic chiasm. This miswiring along the visual pathway affects visual input and processing and results in amblyopia. Normally temporal optic nerve axons do not cross at the optic chiasm, but rather, remain on the original side of the brain. The temporal optic nerve axons in patients with OA, however, irregularly cross over to the other side of the brain. This abnormal number of axons decussating (crossing) the optic chiasm results is detectable via visual evoked potentials.
Interestingly, melanin and pigmented cells appear to play a role in the development of the visual pathway from the eye to the brain, specifically at the optic nerve and chiasmal decussation behind the optic nerve. The reduction or absence of this pigment is believed to result in a misrouting of optic nerve axons at the optic chiasm. This miswiring along the visual pathway affects visual input and processing and results in amblyopia. Normally temporal optic nerve axons do not cross at the optic chiasm, but rather, remain on the original side of the brain. The temporal optic nerve axons in patients with OA, however, irregularly cross over to the other side of the brain. This abnormal number of axons decussating (crossing) the optic chiasm results is detectable via visual evoked potentials.
Similarly, proper foveal development is thought to be dependent on the presence of melanin thus explaining why foveal hypoplasia is commonly observed in ocular albinism. Foveal hypoplasia is a structural underdevelopment of the central portion of the retina that results in reduced photoreceptor function and diminished central visual acuity. Foveal hypoplasia has varying degrees of severity as does ocular albinism. OCT is capable of visualizing the degree of foveal hypoplasia seen as generalized thinness, lack of normal foveal contour, and reduced temporal ganglion cell layer thickness.
The most prevalent congenital abnormalities of the optic nerve leading to impaired vision is optic disc excavation and optic nerve hypoplasia. Excavated optic discs consist of optic disc colobomas, morning glory syndrome, peripapillary staphylomas, and buried optic drusen. Each of these conditions may lead to visual impairment to varying degrees.
Optic nerve hypoplasia describes a small underdeveloped optic nerve which may display the common peripapillary ring (i.e. double ring sign). In addition to abnormal structure, optic nerve hypoplasia often leads to abnormal function of the optic nerve. Each of these optic nerve conditions may be unilateral or bilateral and impair vision mildly or significantly. These conditions may also cause a sensory deprivation accompanied by nystagmus or strabismus.
Furthermore, these ocular conditions may also be linked with systemic conditions, especially neurological or endocrinologic. Therefore multidisciplinary approaches may be necessary if a congenital optic nerve disorder is diagnosed.
One major source of infantile visual impairment is congenital optic nerve disorders. Depending on the condition suspected, neuroimaging or neuro-endocrine workups are warranted. Generally neuroimaging studies are necessitated in cases of congenital bilateral optic atrophy for risk of brain tumors or hydrocephalus while endocrine studies should be performed in cases of optic nerve hypoplasia to evaluate pituitary gland dysfunction. Neuroimaging consists of computed tomography (CT) or magnetic resonance imaging (MRI) and endocrine workups evaluate hormone levels.
 Childhood glaucoma — also termed pediatric glaucoma, infantile glaucoma, or congenital glaucoma — is responsible for approximately 7% of visual impairment in the pediatric population worldwide.
Childhood glaucoma — also termed pediatric glaucoma, infantile glaucoma, or congenital glaucoma — is responsible for approximately 7% of visual impairment in the pediatric population worldwide.
Childhood glaucoma is a collection of diseases with a wide range of clinical progressions and prognoses and is usually diagnosed in the first year of life. Generally speaking, glaucoma is a progressive optic neuropathy, usually associated with elevated intraocular pressure, that initially results first in peripheral vision loss followed by central vision loss if not controlled.
Although relatively rare, childhood glaucoma may result in severe visual impairment or even total blindness if not detected early and properly managed. Childhood glaucoma is classified as either primary childhood glaucoma (PCG) or secondary childhood glaucoma (SCG). PCG is characterized as developmental or naturally occurring while SCG is acquired due to insult to the eye(s).
Childhood glaucoma may occur sporadically, have strong genetic links, be related to systemic disease, or be acquired after a traumatic ocular event. The type of glaucoma the clinical features guides the management and treatment choices.
Understanding the patient history and type of glaucoma it is essential is initiating proper treatment. Secondary childhood glaucoma is oftentimes treated far differently than primary childhood glaucoma. In some cases, genetic testing may be warranted to inform the clinician of an exact diagnosis and allow proper genetic counseling for the family. Accurate glaucoma classification improves patient care, family education, and accurate prognosis. Types of PCGs and SCGs are outlined below.
Primary Childhood Glaucomas (Developmental)
Secondary Childhood Glaucomas (Acquired)
The majority of childhood glaucoma cases are classified at development glaucomas due to developmental defects in the eye(s), particularly in the trabecular meshwork (part of the filtration angle of the eye). 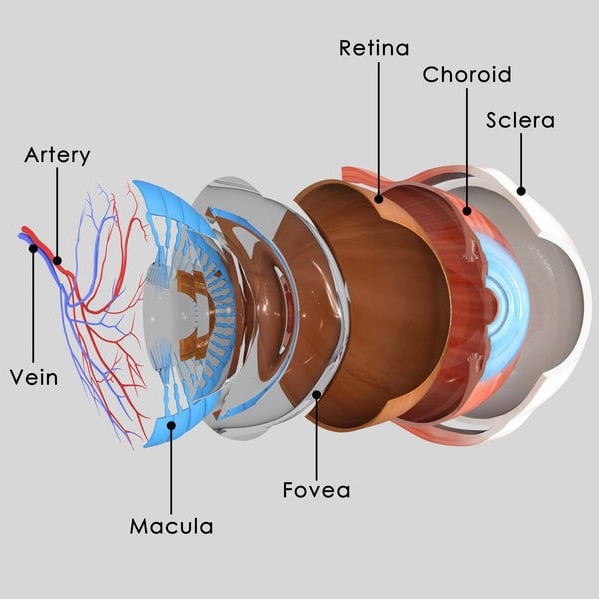 These developmental defects may occur due to genetics, other eye problems, or from systemic disease. Improper drainage of aqueous fluid in the front of the eye leads to a rise in intraocular pressure which may injure the optic nerve due to mechanical pressure or blood flow impairment.
These developmental defects may occur due to genetics, other eye problems, or from systemic disease. Improper drainage of aqueous fluid in the front of the eye leads to a rise in intraocular pressure which may injure the optic nerve due to mechanical pressure or blood flow impairment.
The majority of childhood glaucomas are congenital in nature. Newborn glaucoma is defined as being discovered in the first few months of life, especially because of noticeable anatomical defects in the anterior segment of the eye (front of the eye). Infantile glaucoma is used when the glaucoma is discovered within the two years of life. Late recognized glaucoma describes a congenital glaucoma not discovered until after 2 years of age — these glaucomatous features would have been discovered earlier had an eye examination been performed. When childhood glaucoma occurs as a result of trauma, cataracts, ocular pathological processes, infection, or independent disease processes this is referred to as secondary childhood glaucoma. Secondary childhood glaucoma typically occurs due to impaired function of the drainage angle in the eye.
Glaucoma discovered in childhood is called juvenile glaucoma. As with all glaucomas, the most common subset is open-angle glaucoma. Juvenile open-angle glaucoma (JOAG) is roughly defined as occurring between 5 and 35 years of age and is a form of glaucoma occurring despite the drainage angle of the eye being “open”. The incidence of JOAG is only approximately 1:50,000 people. A diagnosis of JOAG may only be given once secondary glaucomas are ruled out. Unfortunately, due to the unsuspected and insidious nature of JOAG, individuals are often diagnosed late after significant optic nerve damage has ensued, usually partially due to an elevated intraocular pressure. For this reason, and because vision needs to be protected for many more decades, aggressive treatment is often initiated, sometimes surgically.
Genetics and Pathogenesis
When genetically occurring, the inheritance pattern of JOAG is usually due to an autosomal dominant genetic mutation interfering with proteins within the trabecular meshwork (a tissue in the front of the eye responsible for draining aqueous humor). The protein most often disrupted is the myocilin protein (MYOC), previously known as the trabecular meshwork inducible glucose response protein. Over 70 genetic mutations hindering aqueous outflow by impairing the MYOC have been discovered, however not every MYOC mutation always leads to JOAG development.
Diagnosis
Differential diagnosis for JOAG include infectious papillitis, traumatic optic neuropathy, vascular optic neuropathy, steroid induced glaucoma, inflammatory glaucoma, neovascular glaucoma, and congenital glaucomas. Neurologic abnormalities such as Horner’s Syndrome and cranial nerve palsies are not indicative of JOAG.
Management
The vast majority of glaucoma cases worldwide occur in the elderly population where the goal is to preserve vision for the next several decades of life. When a diagnosis of childhood glaucoma is made aggressive treatment often warranted as the goal is to limit vision loss over an entire lifespan. The treatment and management of childhood glaucoma depends on the type of glaucoma diagnosed. JOAG management primarily involves lowering intraocular pressure (IOP) and monitoring any progression through visual field testing and optical coherence tomography scans of the optic nerve and retina. IOP intervention may be initiated through topical medication or surgical treatment. IOP lowering medications primarily focus on either increasing outflow of aqueous humor or decrease the production of aqueous humor. Surgical intervention centers on improving the outflow of aqueous humor from the anterior chamber of the eye. Treatment of JOAG in children is generally fairly aggressive as optic nerve damage is generally thought to be irreversible. 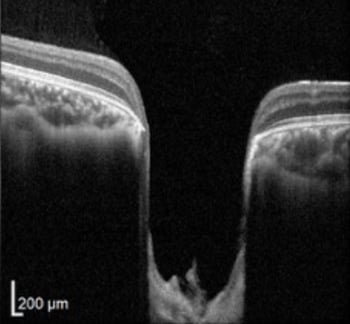
Glaucoma induces an optic neuropathy characterized by axonal death. This is observable during an eye exam as “cupping” or “excavation” of the optic nerve from axon atrophy. Although optic neuropathy generally occurs gradually in adults with glaucoma, damage to the optic nerve may occur rapidly in young children thus necessitating close monitorization and aggressive intervention. There is evidence that cupping may be reversed in children possibly due to the elasticity and developing lamina cribrosa which supports the optic nerve. The goal of treatment for JOAG is lowering IOP, stabilizing IOP, and preserving long-term visual function.
Obstacles to Visual Development
Although the age cutoff is controversial, the critical development period for the eyes and vision is roughly from birth to age 6. The timing of congenital glaucoma interferes with this visual development period. However, conditions such as untreated media opacities causing deprivation immediately from birth have an even higher risk of negatively impacting visual development. Deprivation of vision very early in life causes severe amblyopia as well as poor oculomotor skills and nystagmus (involuntary oscillating eye movements). Impaired visual development results in a condition called amblyopia which forms due to the visual pathway being unable to develop properly due to impaired input and processing of visual information.
Amblyopia risks include media opacities (cataracts, corneal scars), high refractive errors, and strabismus (eye turns). Research shows that the first 6-7 years of life are an especially critical window of time for long-term visual development. If congenital glaucoma, through progressive, presents after the critical development period of the eye — it has a more visually favorable prognosis if well-controlled compared to congenital cataracts or other media opacities.
Conclusion
JOAG is a childhood optic neuropathy usually occurring in both eyes which may cause severe visual impairment or even blindness if not diagnosed early and managed properly. Visual impairment may occur from JOAG from optic neuropathy as well as impaired development of the visual pathway from the eye to the brain. Proper management entails lowering and stabilizing IOP, preventing amblyopia, and preserving long-term visual function by limiting optic neuropathy. JOAG should be closely monitored through regular eye examinations, IOP checks, OCT and visual field testing, and dilated eye examinations. Data from these examinations informs clinicians on the best treatment course for preserving vision and assessing patient outcomes.
Septo-optic dysplasia (SOD), formerly known as de Morsier Syndrome, is characterized by a triad of features: optic nerve hypoplasia, pituitary hormone irregularities, and midline brain defects of the septum pellucidum or corpus callosum.
SOD affects 1:10000 individuals with no gender predilection, but does occur more frequently in children of young mothers. Although clinical features display wide variation the most common findings include optic nerve hypoplasia, hypopituitarism, visual impairment, developmental delays, and seizures.
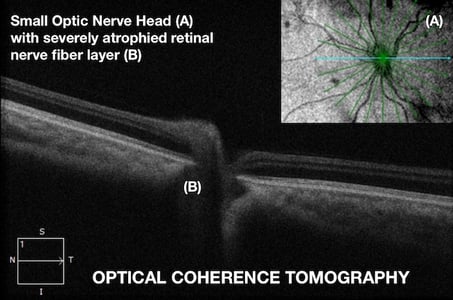 The presence of optic nerve hypoplasia in both eyes, rather than only one eye, increases the risk of developmental delays. Other ocular abnormalities may include strabismus (usually esotropia), nystagmus, microphthalmus, and coloboma. The optic nerve head may display signs of hypoplasia such as an underdeveloped appearance, a double ring sign, or pallor. The degree of visual impairment is highly variable, from near normal to light perception only.
The presence of optic nerve hypoplasia in both eyes, rather than only one eye, increases the risk of developmental delays. Other ocular abnormalities may include strabismus (usually esotropia), nystagmus, microphthalmus, and coloboma. The optic nerve head may display signs of hypoplasia such as an underdeveloped appearance, a double ring sign, or pallor. The degree of visual impairment is highly variable, from near normal to light perception only.
The majority of patients have a best correct vision of around 20/200. One or both eyes may be affected. Visual field testing generally reveals localized defects often in the nasal or inferior fields or overall constriction.
Other clinical features of Septo-optic dysplasia may include:
Midbrain defects classically associated with SOD include agenesis of the septum pellucidum and/or corpus callosum. Abnormalities of the forebrain likely begin in week 4-6 of gestation, a critical developmental period for the anterior neural plate. 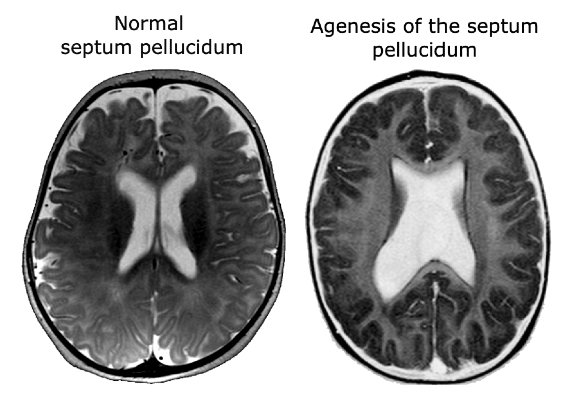 Better understanding of the development of the pituitary gland and the forebrain has led to the discovery of two genes responsible for septo-optic dysplasia, HESX1 and SOX2. However, the role of genetics in SOD is currently limited as genetic abnormalities are detected in less than 1% of cases. This has stirred debate over other causative genes or pathological sources of SOD. A vascular etiology is one proposed mechanism for SOD development as is certain environmental factors such as tobacco, alcohol, or other drugs. More research is needed to fully understand the mechanisms and genetic pathological variants responsible for SOD. Presently most cases are believed to occur sporadically with an absence of family history.
Better understanding of the development of the pituitary gland and the forebrain has led to the discovery of two genes responsible for septo-optic dysplasia, HESX1 and SOX2. However, the role of genetics in SOD is currently limited as genetic abnormalities are detected in less than 1% of cases. This has stirred debate over other causative genes or pathological sources of SOD. A vascular etiology is one proposed mechanism for SOD development as is certain environmental factors such as tobacco, alcohol, or other drugs. More research is needed to fully understand the mechanisms and genetic pathological variants responsible for SOD. Presently most cases are believed to occur sporadically with an absence of family history.
Early diagnosis is vital as this decreases neurodevelopmental burden from hormonal imbalances, notably hypoglycemia and adrenal dysfunction. SOD should be suspected when infants present with jaundice, hypoglycemia, underdeveloped genitals, and nystagmus. In these children baseline endocrinologic workout, neuroimaging, and a comprehensive ophthalmological examination are warranted to confirm or deny SOD.
Septo-optic dysplasia requires a multidisciplinary approach as various organ systems are involved (neurological, ophthalmological, endocrinological). Although genetic research is limited at the moment, genetic counseling is still advisable as the likelihood of family members being affected is increased when a family member is affected.

Cerebral visual impairment (CVI) is a decreased visual response due to a neurological deficit along the visual pathway or the visual center of the brain. Usually, a child with CVI has normal eye findings or an eye condition that cannot explain the degree of visual disturbances. The most common etiology for CVI is perinatal hypoxia, cerebral vascular accident, meningitis, encephalitis, acquired hypoxia, hydrocephalus, or prematurity. Other less common etiologies included intracranial cyst, head trauma, or brain tumors.
Specifically, cortical visual impairment (CVI) denotes vision loss from diminished signaling along the retro-geniculostriate visual pathway (marked E, D, and F) in the absence of other ocular pathology.
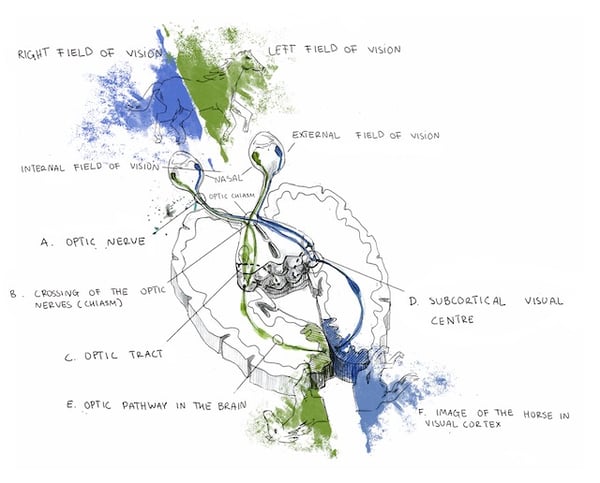 This condition is occasionally more broadly referred to as cerebral, rather than cortical in nature, as reduced vision may occur due to abnormalities in the subcortical optic radiations or occipital cortex. The incidence of CVI has been steadily rising over the past few decades and currently around 160 cases per 100,000 individuals. Worldwide, CVI is a major contributor to childhood vision loss. Visual function is diminished in CVI due to reduced or absent signaling from the eye to the brain. The degree of visual impairment ranges from mild to complete vision loss, called cortical blindness.
This condition is occasionally more broadly referred to as cerebral, rather than cortical in nature, as reduced vision may occur due to abnormalities in the subcortical optic radiations or occipital cortex. The incidence of CVI has been steadily rising over the past few decades and currently around 160 cases per 100,000 individuals. Worldwide, CVI is a major contributor to childhood vision loss. Visual function is diminished in CVI due to reduced or absent signaling from the eye to the brain. The degree of visual impairment ranges from mild to complete vision loss, called cortical blindness.
Children with CVI present with reduced vision, normal pupil responses, no nystagmus, and relatively normal ocular findings, although optic disc pallor may present later (quite rare). These children oftentimes have hyperopic (farsighted) refractive errors, strabismus (eye turns), and visual field defects (usually bilateral inferior defects or homonymous hemianopia). One unique feature of CVI sometimes observed is dyskinetic strabismus where an eye turn may switch between exotropia (outward) and esotropia (inward) sporadically. While nystagmus is absent in cases of cortical damage, a nystagmus may be present if the damage is subcortical at the level of the optic radiations.
CVI has many causes, but the most common in children is a perinatal hypoxic episode (asphyxia). Location of ischemic damage is dependent on infant age and term (premature vs full-term) due to the location of the cerebral watersheds at that developmental time. In preterm infants, infarctions occur in the periventricular deep white matter causing periventricular leukomalacia and affecting the optic radiations. Parasagittal infarctions are rare in preterm infants due to bridging of the meningeal anastomosis between the watershed zone and major cerebral arteries. In full-term infants, the watershed areas lie in the regions between the anterior and middle cerebral arteries as well as the medial and posterior cerebral arteries. At this stage of development, hypoxia-induced hypoperfusion to the watershed areas leads to infarction to the frontal, temporal, parietal, and occipital brain areas. Generally, insult to the optic radiations, rather than the cortical areas, has a worse prognosis. Ischemic brain damage may occur before or after birth, delaying dendrite formation and synaptogenesis, causing abnormal myelination of visual pathways, or leading to cell death of glial and neural cells due to impaired protein synthesis. Improved visual function in children with CVI may be due to rerouting of axons, reduced axon retraction, and reactive synaptogenesis.
Although most cases of CVI occur due to perinatal hypoxia, ischemic damage during infancy or later may also cause CVI. Postnatal hypoxia-ischemic brain injury along the visual pathway may occur due to cardiac arrest, respiratory distress, generalized hypotension, hypertensive crisis induced cerebral occlusion, transtentorial herniation compressing cerebral arteries, cardiac surgery, or air embolisms. Vascular malformations or central nervous system tumors may compress cranial vessels causing CVI. Congenital cyanotic heart disease may cause emboli formation and thrombotic disorders may arise from trauma, meningitis, polycythemia, neurofibromatosis, and sickle cell disease. Any number of conditions inducing cerebral ischemic injury may cause CVI during infancy.
CVI may occur from ischemic damage due to intracranial hemorrhages, which commonly occur in premature infants within the first few days of life. Such hemorrhages occur either spontaneously or from a hypoxic crisis in small vessels within the ventricular walls due to inadequate vascular support. Following an ischemic incident, reperfusion of these weak blood vessels may cause a hemorrhage. Hydrocephalus may ensue from these hemorrhages, dissecting the brain parenchyma and inflicting damage to the visual pathways. Also common in preterm infants are choroidal plexus hemorrhages which may cause subsequent periventricular leukomalacia and CVI.
CVI and congenital homonymous hemianopia may occur due to a multitude of cerebral malformations including: occipital encephaloceles, parietal encephaloceles, hydranencephaly, Chiari malformations, Dandy-Walker complex, porencephalic cysts, or neuronal migration abnormalities. Porencephalic cysts may occur from vascular compromise, infections, or hemorrhagic dissection while neuronal migration abnormalities may occur due to infections, ischemia, or metabolic disturbances. Migrational abnormalities result in normal neurons in abnormal locations. Clinical manifestations of these anomalies depend on the extent, timing, and location of migration which cause different categories of migrational abnormalities. The most severe migrational abnormality is lissencephaly which includes agyria and pachygyria. Other categories include neuronal heterotopias, unilateral megalencephaly, schizencephaly, porencephaly, and encephalomalacia.
 Solid brain tumors develop in approximately 30 out of 1,000,000 children and have a 10 year survival rate of just under 60%. Diagnosis is made through both clinical history and neuroradiological imaging and confirmed through histology. Despite the rare diagnosis and survival rate, brain tumors are the leading cause of cancer deaths in children. These tumors can cause significant visual impairments if the tumor is located in areas of the visual system or if the tumor leads to hydrocephalus (elevated intracranial pressure). Such visual impairments may negatively affect a childs everyday activities, development, education, and vocation if not treated effectively or in a timely manner.
Solid brain tumors develop in approximately 30 out of 1,000,000 children and have a 10 year survival rate of just under 60%. Diagnosis is made through both clinical history and neuroradiological imaging and confirmed through histology. Despite the rare diagnosis and survival rate, brain tumors are the leading cause of cancer deaths in children. These tumors can cause significant visual impairments if the tumor is located in areas of the visual system or if the tumor leads to hydrocephalus (elevated intracranial pressure). Such visual impairments may negatively affect a childs everyday activities, development, education, and vocation if not treated effectively or in a timely manner.
Symptoms of Brain Tumors in Children
Due to the brain tissue already fitting tightly inside the enclosed skull, tumor growth causes increased pressure within the skull, termed elevated intracranial pressure or hydrocephalus. Elevated intracranial pressure occurs mostly due to excess cerebrospinal fluid. This excess pressure exerted on the surrounding brain tissue, if severe, causes neurological dysfunction manifesting as disturbed vision, poor balance and coordination, impaired motor function, headaches, cognitive impairments, nausea and vomiting, irritability, drowsiness, or seizures. Symptoms are dependent on the location and size of the tumor as well as the degree of elevated intracranial pressure.
Brain Tumor Diagnosis in Children
Symptoms of brain tumors which locate close to visual pathways or visual centers often manifest initially as vision problems. For this reason, eye care professionals may be the first healthcare providers notified of tumor-related symptoms. Although not always, signs of brain tumor formation may be visible in the eye as optic nerve edema. Regardless of the presence of ocular signs, children experiencing brain tumor symptoms should be thoroughly and immediately evaluated by a pediatric neurologist, pediatrician, or emergency room physician.
 Neurological examination and neuroimaging are the first steps taken to confirm or deny the presence of an intracranial process as a tumor. Multiple scan types may be necessary to confirm the presence or absence of a tumor depending on the size, location, and type of tumor. Common neuroimaging methods include:
Neurological examination and neuroimaging are the first steps taken to confirm or deny the presence of an intracranial process as a tumor. Multiple scan types may be necessary to confirm the presence or absence of a tumor depending on the size, location, and type of tumor. Common neuroimaging methods include:
Brain tumors are classified by a standard created by the World Health Organization. This classification is characterized by the tumor cell type and location. Tumor types are differentiated by the cell type involved and the tumor grade represents the aggressiveness of the cells. Tumor grade ranges from 1-4 and is usually denoted in Roman numerals. The tumor grade is essential for treatment determination and prognosis predictions. High grade tumors (III and IV), grow quicker, inflict more damage, are more difficult to treat, and have worse survival rates than low grade tumors (I and II). High grade tumors are considered more malignant or cancerous. Conversely, low grade tumors progress slowly, inflict minimal damage, and have high long-term survival rates. In tumors, the most malignant cell present determines the grade for the tumor as a whole. Certain tumors may become malignant over time and change growth patterns.
Types of Brain Tumors in Children
Primary brain tumors (PBTs) may be subdivided into three groups: medulloblastomas, ependymomas, and astrocytomas. This classification is determined by the tissue origin, location, and histopathological characteristics. Medulloblastomas, accounting for approximately 20% of PBTs, are the most common malignant childhood brain tumors and are neuroectodermal tumors of the cerebellum. Ependymomas are the third most common PBTs of the CNS and oftentimes arise in the parenchyma of the cerebral hemispheres. Astrocytomas account for roughly 50% of all brain tumors in children and are the most common type of glioma. Four main types of astrocytomas exist in children:
Other Pediatric Brain Tumors
Brain stem gliomas: The majority of these tumors are located in the middle of the brainstem making them inoperable and difficult to treat.
Choroid plexus tumors: These tumors develop in the choroid plexus cells in the ventricles of the brain. These cells are responsible for producing cerebrospinal fluid which cushions the brain and spinal cord. This type of tumor may result in hydrocephalus, or a buildup of cerebrospinal fluid.
Craniopharyngiomas: These benign tumors are located near the pituitary gland, responsible for producing hormones controlling other glands in the body. Tumors in this area may cause hormonal imbalances. Surgical removal is usually the preferred method of treatment.
Germ cell tumors: These tumors arise from germ cells (eggs or sperm) during development and may be benign or malignant.
Medulloblastomas: These brain tumors account for approximately 15% of pediatric brain tumors and usually develop between the ages of 4-9 and disproportionately more in males. These tumors are malignant and may metastasize to the spinal cord. Treatment typically is a combination of surgery and other methods.
Optic nerve gliomas: This is a glioma found in or surrounding the optic nerves. These tumors are more frequently discovered in children with neurofibromatosis, a genetic disorder or the nervous system. Optic nerve gliomas cause impaired vision and hormonal problems due to the location. Treatment can be difficult due to sensitive surrounding brain structures.
When planning for a pediatric brain tumor surgery, the neurosurgery team needs to know the tumor:
Treatment for Pediatric Brain Tumors
Surgery: The majority of childhood brain tumors necessitate surgical removal. The initial surgery may entail complete removal or partial removal to alleviate elevated intracranial pressure. Surgical removal may be the only treatment necessary for low-grade tumors.
Follow-Up Care: Future treatment and recovery process of the child after surgery is largely dependent on the type, location, and size of the tumor as well as the timing of diagnosis and treatment. The prognosis is highly variable and case dependent. Regular post-operative and follow-up visits are needed to monitor neurological status, side effects, progression, or recurrence.
Radiation Therapy consists of focusing high doses of radiation directly on the tumor itself and surrounding tissue. In infants and young children, radiation is used sparingly as the brain is still in critical development. However certain tumors, such as medulloblastomas, may require radiation to the entire brain or spinal cord.
Chemotherapy utilizes anti-cancer chemicals systemically for high-grade, aggressive tumors. Chemotherapy may be administered in the form of pills (orally), intravenously (vein), or injection (cerebrospinal fluid or brain cavity after surgical removal).
 The term head trauma is exceedingly broad and may range from minor symptoms to severe and life-threatening complications. Traumatic head injury may cause concussions, contusions, lacerations, or brain damage. This may clinically manifest as loss of consciousness (from second to hours) or, in severe cases, leads to coma. Head trauma may also result in further complications such as hemorrhages in the epidural, subdural, subarachnoid, or intracerebral areas.
The term head trauma is exceedingly broad and may range from minor symptoms to severe and life-threatening complications. Traumatic head injury may cause concussions, contusions, lacerations, or brain damage. This may clinically manifest as loss of consciousness (from second to hours) or, in severe cases, leads to coma. Head trauma may also result in further complications such as hemorrhages in the epidural, subdural, subarachnoid, or intracerebral areas.
Well known that visual impairment is frequently associated with head trauma and typically occurs immediately thereafter. Vision lost due to significant head trauma is sometimes referred to as cortical visual impairment (CVI). CVI does not exclusively occur due to head trauma, but is a general term describing vision loss due to impaired neurological signaling along the visual pathway in the brain. The long-term effects of CVI vary significantly ” ranging from total recovery of the eyesight within days to permanent vision loss. Understandably, more severe cases of head trauma are associated with a worse prognosis when compared to minor head trauma cases. Patients afflicted with severe head trauma frequently have long-term visual sequelae and permanent neurological deficits. These cases typically are evident through neuroimaging, revealing skull fractures, intracranial hemorrhages, or cerebral injury.
While after a traumatic event certain symptoms do typically improve quickly ” such as headaches, irritability, blindness, light sensitivity, disorientation ” other symptoms such as cognitive deficits and visual field defects may persist due to posttraumatic injury and /or atrophy of brain tissue. Common complications arising after head trauma include hydrocephalus, cerebral edema, and hemorrhages. Cerebral dysfunction due to head trauma may also result in concussions, epilepsy, brain tissue edema, or ischemia.
Common visual symptoms of traumatic brain damage may include blurred central vision, missing peripheral vision, light sensitivity, spatial orientation difficulty, reduced peripheral awareness, double vision, or fine flickering of vision resembling TV static (visual snow). Sometimes neuroimaging results are negative as certain neurological disturbances, like concussions, are functional rather than structural in nature.
Young children often are unable to recognize vision loss or vocalize visual disturbances, however these children may exhibit signs of irritability, uncooperativeness, restlessness, confusion, disorientation, lack of coordination, headaches, or drowsiness. Therefore, trauma induced CVI should be suspected in children exhibiting such behavior as this may be a psychological reaction to either the brain trauma or visual disturbances. Visual disturbances may arise due to damage of the posterior visual pathway likely from the coup or contrecoup forces from the trauma. Transient blindness potentially occurs due to local cerebral vasospasm similar to the pathophysiology of migraines. Similarly, visual snow symptoms have been associated with migraines and may have a vascular etiology. Persons with a family history of migraines seem to be at increased risk for transient vision loss following a traumatic brain event. Certain persons may be predisposed to these vascular disturbances following head trauma, as evidenced by certain people repeatedly having such transient vision loss following striking a soccer ball with their head on multiple occasions (aka œfootballer's migraine). This and other cases suggests certain persons, likely with a family history of migraines, are at increased risk of visual disturbances due to cerebral blood flow alterations following even minor head trauma.
Sadly, CVI may also occur due to child abuse and shaken baby syndrome. Such trauma may induce intracranial hemorrhages, concussive cerebral injury, subdural hematomas, or retinal hemorrhages from a young age. Such diagnoses warrant consideration when these neurological signs are present along with signs of physical trauma.
Significant metabolic disturbances may lead to not only optic neuropathy, but also cortical visual impairment (CVI). These disturbances could be due to extreme hypoglycemia, poisoning (carbon monoxide, lead, nitric oxide), uremia, hemodialysis, alcohol, or cocaine exposure. CVI is also associated with neurodegenerative disorders such as metabolic encephalopathy, lactic acidosis, mitochondrial encephalopathy, Leighs disease, X-linked adrenoleukodystrophy, and strokes.
A small number of CVI cases are caused by pathogens. Roughly 5% of CVI results from bacterial meningitis during infancy. Meningitis has well-known neurological complications such as cognitive deficits, seizures, hemiplegia, and quadriplegia. However, meningitis is also associated with significant visual impairment, visual field defects, and visual hallucinations. The most likely organisms are haemophilus influenzae, pneumococci, and streptococci. Meningitis induced CVI occurs within 1 week for 50% of cases and within 1 month for essentially all cases. For unknown reasons, haemophilus influenzae seems to target the occipital cortex, largely responsible for vision. Post-meningitic CVI may be a consequence of hydrocephalus, ischemia, venous sinus thrombosis, or thrombophlebitis. As with CVI in general, post-meningitic CVI may have partial recovery, complete recovery, or permanent vision loss.
Viral encephalitis caused by herpes simplex virus is associated with severe CVI with the vast majority (~80%) of these cases being caused by Type 2 herpes simplex. These cases usually involve severe brain damage from necrotizing encephalopathy, widespread neurological disease, and demyelination causing diffuse white matter damage. Severe cortical damage and optic atrophy are both frequently associated with neonatal herpes simplex virus, leading to profound vision loss.
Hydrocephalus, or buildup of excessive cerebrospinal fluid in the ventricles of the brain, exerts pressure on the brain which may cause visual impairment. Hydrocephalus may arise from multiple disease processes including meningitis, space occupying lesions, enlarged brain ventricles, brain malformation. Hydrocephalic brain damage and dysfunction is multifaceted.
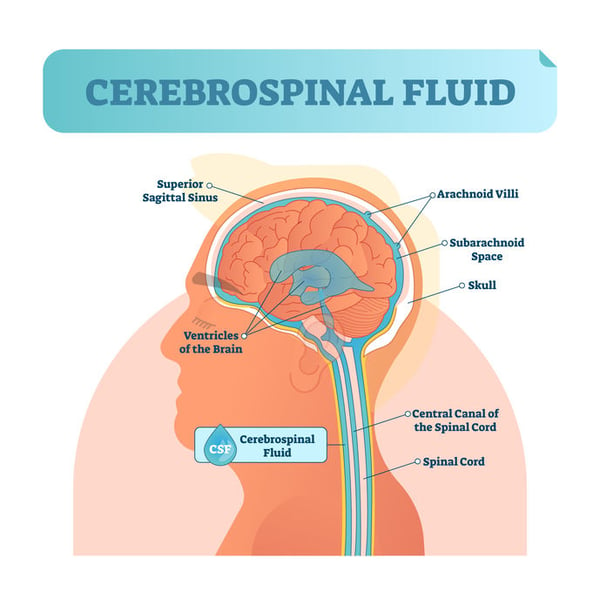
Hydrocephalus may cause axonal dysfunction through mechanical stretching or compression of tissue as well as from ischemia due to impaired blood flow, especially in white matter. Inflammation and oxidative stress may further deteriorate axons. Extracellular fluid dynamics might also be altered due to excess cerebrospinal fluid.
Visual impairment from hydrocephalus may present as visual field defects, the most common being homonymous hemianopia, or loss of vision to the right or to the left. Hydrocephalus may cause damage to either the anterior or posterior visual pathway. Anterior visual pathway damage may occur from papilledema-induced optic atrophy, compression of the optic chiasm from the third ventricle, compression of the optic tracts, developmental abnormalities, or vascular deficiencies causing ischemia. Posterior visual pathway damage may result from compression of the posterior cerebral arteries potentially causing laminar necrosis in the visual cortex. Although hydrocephalus may cause a variety of neurological problems, the visual system is commonly affected. In children vision problems manifest as reduced visual acuity, visual field defects, strabismus, eye movement disorders, and impaired orientation and depth perception.
Treatment of hydrocephalus, oftentimes using a shunt, may partially or completely resolve visual field defects. If hydrocephalus reoccurs after a shunt procedure, a shunt revision may be necessary. Vision disturbance may be the first or only sign of a shunt failure. Vision generally improves over the course of several years, although may improve as quickly as a few days.
Fedorov Restoration Therapy is a painless, straightforward therapeutic process developed to improve the function of partially damaged or degenerated retinal cells. Weak or absent retinal signaling limits the flow of information from the eye to the brain, causing a variety of ocular symptoms and eventually leading to lost or impaired vision.

Our therapeutic approach involves the application of weak electrical currents designed to indirectly stimulate retinal cells. This electrical activitaton enhances the activity and function of these retinal cells thereby reinforcing visual signaling along the optic nerve and partially restoring impaired eyesight.
Fedorov Restore Vision Clinic provides outpatient treatment where patients attend the clinic daily (excluding weekends) over a two week period for therapeutic sessions lasting approximately two hours per day.
Patients undergo comprehensive diagnostic testing in order to determine the degree of vision loss and to establish baseline parameters by which post-therapy results can be measured. Patients will have subjective vision evaluations (visual acuity, contrast vision, color vision), detailed visual field assessment, advanced OCT imaging of optic nerve and retinal structures, and electrophysiological testing. After these tests are conducted, the data is analyzed and interpreted by physicians. Patients are then ready for the treatment to be administered.
Electrostimulation therapy is administered by alternating electrical currents through electrodes attached around the eyes. During the session patients generally experience a sense of visual excitement, and will see (what appears to be) lights (this is called the “phosphene effect”).
Once treatment is complete, patients undergo the same tests conducted at the preliminary visit. After reassessment the treatment results will be thoroughly discussed during the final visit. Ultimately, the major goal of Fedorov Therapy is not only to restore visual function but to improve quality of life for our patients. In fact, pre and post therapy surveys reveal that the vast majority of our patients report an improvement in everyday activities, reclaimed social functioning, enhanced independence, and an overall higher quality of life. If you or a loved one are experiencing vision problems, especially caused by retinal or optic nerve damage, Fedorov Restoration Treatment might be the right treatment for you.
 Note: The information given in this blog are the opinions of the authors and for reader familiarization purposes only. This blog is not intended as a substitute for professional medical advice. Also, the information provided does not replace or abolish any official or legal terms for diagnosis, treatment, and management. Authors are not liable for any undesirable consequences or effects related to the information provided in the blog.
Note: The information given in this blog are the opinions of the authors and for reader familiarization purposes only. This blog is not intended as a substitute for professional medical advice. Also, the information provided does not replace or abolish any official or legal terms for diagnosis, treatment, and management. Authors are not liable for any undesirable consequences or effects related to the information provided in the blog.
This page prepared in cooperation with Kaleb Abbott, O.D., M.S.


.jpg?width=500&name=Fotos%20-%201%20von%201%20(1).jpg)