.jpeg?width=1280&name=Importe%20-%201%20von%201%20(1).jpeg) The retina is a very thin layer of cells covering the posterior (back) pole of the eyeball that captures external light, converts this light into neural signals, and sends these signals via the optic pathways to the brain. Fascinatingly, the retina is only approximately 0.5mm thick – three sheets of computer paper – yet is the tissue responsible for showing us the world by sending electrical signals to the brain throughout our lives. When functioning properly the human retina is capable of resolving approximately 576 megapixels. Compare that with the latest iPhone with its touted camera of 12 megapixels. Artificial intelligence and camera lenses remain light-years away from reaching the impeccable clarity of the human retina.
The retina is a very thin layer of cells covering the posterior (back) pole of the eyeball that captures external light, converts this light into neural signals, and sends these signals via the optic pathways to the brain. Fascinatingly, the retina is only approximately 0.5mm thick – three sheets of computer paper – yet is the tissue responsible for showing us the world by sending electrical signals to the brain throughout our lives. When functioning properly the human retina is capable of resolving approximately 576 megapixels. Compare that with the latest iPhone with its touted camera of 12 megapixels. Artificial intelligence and camera lenses remain light-years away from reaching the impeccable clarity of the human retina.
Despite its thinness, the retina is composed of 10 distinct layers. Light travels through the thickness of the retina before stimulating photopigment inside the photoreceptors (specialized cells called cones and rods). These photoreceptors in the posterior (back) retina photo-chemically convert photons of light into neural signals through a process called phototransduction. These signals travel from the posterior (outer) to the anterior (inner) retina and are modified and integrated along the way by various cells in other retinal layers. The photochemical transduction process is tremendously complicated and still not fully understood. Phototransduction requires enormous amounts of oxygen and nutrients.
 In fact, the retina is known to be the most metabolically active tissue in the body and photoreceptors require more oxygen than any other human cell. The combination of photoreceptors demanding a massive oxygen supply with the relatively low vascular nature of the outer retina substantially increases the risk of cell damage by hypoxic injury. This delicate, transparent tissue of exceedingly complicated anatomy also requires substantial metabolic support, which predisposes the retina to dysfunction with even the slightest abnormalities.
In fact, the retina is known to be the most metabolically active tissue in the body and photoreceptors require more oxygen than any other human cell. The combination of photoreceptors demanding a massive oxygen supply with the relatively low vascular nature of the outer retina substantially increases the risk of cell damage by hypoxic injury. This delicate, transparent tissue of exceedingly complicated anatomy also requires substantial metabolic support, which predisposes the retina to dysfunction with even the slightest abnormalities.
The process of creating, using, disposing, and regenerating metabolites used in phototransduction is part of the visual cycle. Small retinal or metabolic anomalies may disrupt the visual cycle and the phototransduction process thereby hindering these electrical signals from reaching the brain via the optic nerve. Inhibition of any part of this process may result in visual impairment. This may be a general reduction of vision, vision loss in specific areas of the retina (and therefore field of vision), or diminished types of functional vision such as color or contrast vision.
As mentioned above, despite the astonishing thinness of the retina, it is composed of 10 distinct layers. Adequate phototransduction and sufficient conveyance of visual signals is accomplished through highly specialized retinal anatomy. The retina is shaped like a circular disc between 30-40mm in diameter, with the center 6mm being considered the central retina (macula). Only the macula, which accounts for 3-5% of the total retina, is capable of achieving 20/20 vision. The remaining retina has far less image resolution capability.
A protective yellow-pigmented layer consisting of xanthophyll carotenoids (lutein and zeaxanthin) called the macula lutea covers the macula and filters out short-wavelength (blue) light. The centermost part of the macula, termed the fovea, is responsible for sharp, central vision. An anatomical pit is located at the center of the fovea giving the retina its thinnest area at only approximately 200-220 microns. This foveal contour occurs due to retinal layers being displaced laterally. Several retinal layers disappear in the fovea making room for densely packed cone-photoreceptors, which enhance visual sharpness (image). Interesting, the fovea is avascular (it has no direct blood vessels to supply cells). With the exception of the foveal pit, the macula is generally thicker than the peripheral retina due to increased density of cone photoreceptors.
The most peripheral portion of the retina is called the ora serrata, radially located approximately 21mm from the fovea. Photoreceptor-wise, cones dominate the central retina while rods are found in the peripheral retina. The neural-retina oftentimes refers to the first 9 layers of the retina, while the retinal pigment epithelium (RPE) is considered a supportive layer for the neural-retina. The RPE is not involved in photochemical conversion of light into electrical signals, but rather, protects and supports the neural-retina, especially through the blood-retinal barrier. The retina is transparent in nature to allow for light to pass through the anterior (front) retinal layers before reaching the photoreceptors in the posterior (back) retina. The architecture of the human retina demonstrates remarkably clear anatomical and functional distinction.
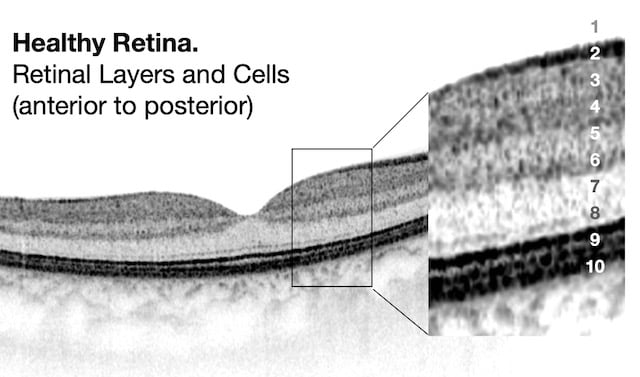
OCT Abbreviation:
1. Internal limiting membrane (ILM) – footplate of Muller cells
2. Retinal nerve fiber layer (RNFL) – axons of ganglion cells
3. Ganglion cell layer (GCL) – nuclei of ganglion cells
4. Inner plexiform layer (IPL) – synapse between bipolar cells with ganglion and amacrine cells
5. Inner nuclear layer (INL) – nuclei of amacrine, bipolar, and horizontal cells
6. Outer plexiform layer (OPL) – synapses between rods/cones with bipolar and horizontal cells
7. Outer nuclear layer (ONL) – cells bodies (nuclei) of rods and cones
8. External limiting membrane (ELM) – Muller cell endplates
9. Photoreceptor layer (PL) – contains outer segments of rods and cones
10. Retinal pigment epithelium (RPE) – nourishes and supports neural-retina, absorbs stray light with pigment
The human visual system is a remarkably intricate arrangement beginning with allowing external light stimuli to project through the retina and be subsequently absorbed by photopigments within the photoreceptors. These photons are transduced into a biochemical message before becoming an electrical signal that is modulated by ensuing retinal cells as this neural signal travels to the anterior (front) of the retina and then to the optic nerve via retinal ganglion cell axons. The optic nerve is the part of the visual pathway that connects retinal cells with the brain. In the primary visual cortex of the brain (left and right occipital lobes) (image), this neural retinal signal is processed and analyzed, completing the transformation of previous photon light signals into detailed and practical visual information.
Research indicates this visual signal is processed and refined as it travels through the retina to the visual cortex. Some of this modulation occurs via a vertical excitatory pathway of the retina, while other alterations transpire through two lateral inhibitory pathways. Horizontal cells in the outer retina supply inhibitory feedback of the photoreceptors while amacrine cells of the inner retina deliver inhibitory feedback of the bipolar and ganglion cells. Here is a brief summary of the functions of specific retinal cell types.
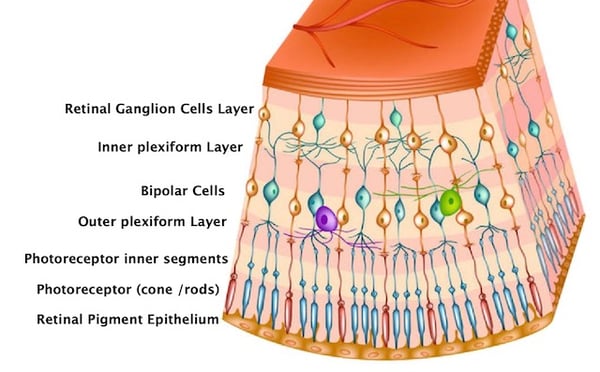
Rods and cones: capture and phototransduce light into bioelectrical signals
Bipolar: receive signals from photoreceptors and transmit signals to the inner retina
Ganglion*: transmit neural signals to the brain via the optic nerve
Horizontal: modulate and integrate neural signals, lateral inhibition to OPL
Amacrine: modulate and integrate neural signals, lateral inhibition to IPL
Interplexiform: modulate and integrate neural signals, lateral inhibition to OPL and IPL
Muller: structure and functional support
*a subtype of retinal ganglion cells called intrinsically photosensitive retinal ganglion cells (ipRGCs) are a third type of photoreceptor which are stimulated by blue light and modulate circadian rhythm, pupil responses, migraines, and certain moods.
As previously mentioned, the retina is one of the most metabolically active tissues in the human body and requires tremendous oxygen consumption to properly function. This metabolically demanding tissue requires an extensive, dual vascular supplying network of vessels which both originate from the ophthalmic artery.
-2.jpeg?width=500&name=Importe%20-%201%20von%201%20(2)-2.jpeg)
Eye fundus image. Both source of retinal blood supply are seen.
Note: Patient has severe atrophy of choroid - it's why choroidal vessels well seen.
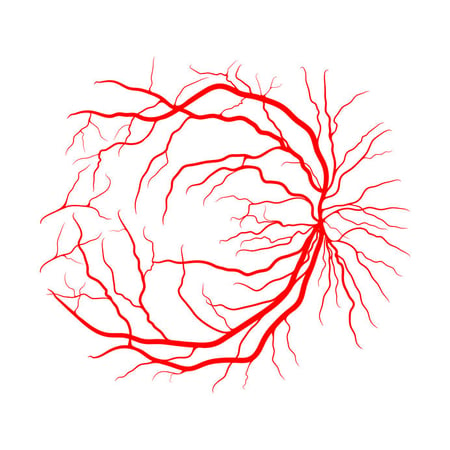
Only the inner portion of the retina is actually supplied by retinal blood vessels. This anterior two-thirds of the retina receives its blood supply from the central retinal branch of the ophthalmic artery. This central retinal artery enters along the optic nerve and branches into four distinct arterial arcades at anterior surface of the retina, which are observable via ophthalmoscopy. These arteries branch into arterioles traveling through the RNFL whose shadows are perceptible using optical coherence tomography (OCT) imaging. These arterioles form an interconnecting plexus of three sets of capillaries between the inner RNFL, outer RNFL and GCL, and the INL. Despite only accounting for two-thirds of the thin retinal tissue, the inner retina receives 20-30% of total ocular blood flow. Although this blood supply is primarily for the inner retina, oxygen may extend as far as the inner photoreceptors during times of intense oxygen demand. A similar and parallel system of venules and veins exists for drainage.
-3.jpeg?width=450&name=Importe%20-%201%20von%201%20(1)-3.jpeg) The outer portion of the retina is supplied through the choroid and not through retinal circulation (image). This posterior third of the retina is supplied through a “large-capillary lake” layer called the choriocapillaris, which is located directly behind the retina and is fed by the choroid. By unit weight, the choroid has the greatest blood flow of any tissue in the human body, the highest perfusion rate of any vascular bed, and receives 65-85% of total ocular blood flow. The choroid primarily receives blood supply from the long and short ciliary branches of the ophthalmic artery, but the anterior ciliary arteries contribute to a lesser degree. In addition to supplying the outer third of the retina, the choroid also supplies the RPE and part of the optic nerve head. Since the outer retina is avascular, the choroid provides oxygen and nutrients to the photoreceptors through free diffusion. The one or two vortex veins in each retinal quadrant drain the choroidal blood.
The outer portion of the retina is supplied through the choroid and not through retinal circulation (image). This posterior third of the retina is supplied through a “large-capillary lake” layer called the choriocapillaris, which is located directly behind the retina and is fed by the choroid. By unit weight, the choroid has the greatest blood flow of any tissue in the human body, the highest perfusion rate of any vascular bed, and receives 65-85% of total ocular blood flow. The choroid primarily receives blood supply from the long and short ciliary branches of the ophthalmic artery, but the anterior ciliary arteries contribute to a lesser degree. In addition to supplying the outer third of the retina, the choroid also supplies the RPE and part of the optic nerve head. Since the outer retina is avascular, the choroid provides oxygen and nutrients to the photoreceptors through free diffusion. The one or two vortex veins in each retinal quadrant drain the choroidal blood.
 The health of the retinal blood supply can be evaluated using several different ophthalmological instruments. Fundus photography provides similar views of the retinal blood supply as dilated fundus examination, but in two dimensions rather than three. These images display the retinal architecture supplying blood flow to the inner retina, but not clear views of the underlying choroid (image). Optical coherence tomography (OCT) provides cross-sectional scans of the retinal layers and choroid. Although not particularly helpful in visualizing specific retinal vessel architecture, the aftermath of unhealthy retinal vessels is quite apparent, such as with fluid edema or new blood vessel growth (neovascularization). OCT angiography (OCTA) on the other hand is a new, powerful tool for analyzing retinal and choroidal intravascular blood flow in real time that may reveal vessel leakage, capillary dropout, or ischemia. Angiography (FA) is the traditional method of assessing retinal and choroidal circulation during different stages of flow. Although being extremely effective in diagnosing circulation abnormalities, FA is accompanied with certain risks such as nausea, vomiting, headaches, fatigue, and even anaphylaxis. OCTA is not a complete substitute for FA, but OCTA does provide a quick, painless, and non-invasive alternative to traditional FA in certain cases.
The health of the retinal blood supply can be evaluated using several different ophthalmological instruments. Fundus photography provides similar views of the retinal blood supply as dilated fundus examination, but in two dimensions rather than three. These images display the retinal architecture supplying blood flow to the inner retina, but not clear views of the underlying choroid (image). Optical coherence tomography (OCT) provides cross-sectional scans of the retinal layers and choroid. Although not particularly helpful in visualizing specific retinal vessel architecture, the aftermath of unhealthy retinal vessels is quite apparent, such as with fluid edema or new blood vessel growth (neovascularization). OCT angiography (OCTA) on the other hand is a new, powerful tool for analyzing retinal and choroidal intravascular blood flow in real time that may reveal vessel leakage, capillary dropout, or ischemia. Angiography (FA) is the traditional method of assessing retinal and choroidal circulation during different stages of flow. Although being extremely effective in diagnosing circulation abnormalities, FA is accompanied with certain risks such as nausea, vomiting, headaches, fatigue, and even anaphylaxis. OCTA is not a complete substitute for FA, but OCTA does provide a quick, painless, and non-invasive alternative to traditional FA in certain cases.
The brain and retina exhibit anatomical, functional, and immunological similarities that may explain the link between retinal and neurological degeneration. 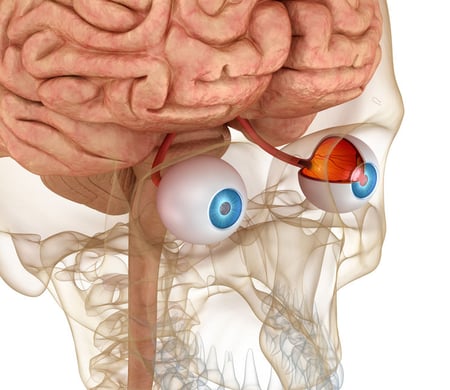
Evidence is now emerging suggesting associations between retinal atrophy and brain atrophy of the primary visual cortex. These findings demonstrate structural and functional alterations within the retina and brain for certain retinal and neurological conditions. Furthermore, retinal input may elicit neurodevelopmental reorganization in the primary visual cortex. For instance, Leber Hereditary Optic Neuropathy – known to cause retinal ganglion cell death – has been associated with reduced gray matter in the primary visual cortex of the brain. Glaucoma and age-related macular degeneration may also have corresponding neurological degeneration. Conversely, retinal changes have been observed in patients with Parkinson’s, Alzheimer’s, multiple sclerosis, and even strokes.
In every facet, the retinal proves to be notoriously complicated – intricate retinal architecture, elaborate cellular function, convoluted blood supply, and even associations with neurological degeneration and function. Sharp visual function is dependent on immaculate retinal structure and copious oxygen and nutrient availability. For this reason, visual function may be disturbed with even the slightest retinal irregularity. Retinal pathology is defined by which abnormality occurs, the subsequent retinal alteration, and the ensuing visual changes.
The course and progression of RP has extensive variation amongst individuals predominantly due to the genetic heterogeneity involved. Modes of inheritance include autosomal dominant (AD), autosomal recessive (AR), X-linked (XL), and mitochondrial with AD, AR, and XL being the most common. Genetic testing and counseling is recommended with RP to better inform prognosis, therapeutic approaches, and potential family involvement. Non-syndromic RP is linked with up to 150 mutation variants involving 50 different loci and 84 different genes. The number of loci and genes associated with each inheritance mode are – 26 loci and 21 genes for ARRP, 18 loci and 17 genes for ADRP, and 6 loci and 2 genes for XLRP. The number of associated genes is steadily increasing as molecular testing advances.
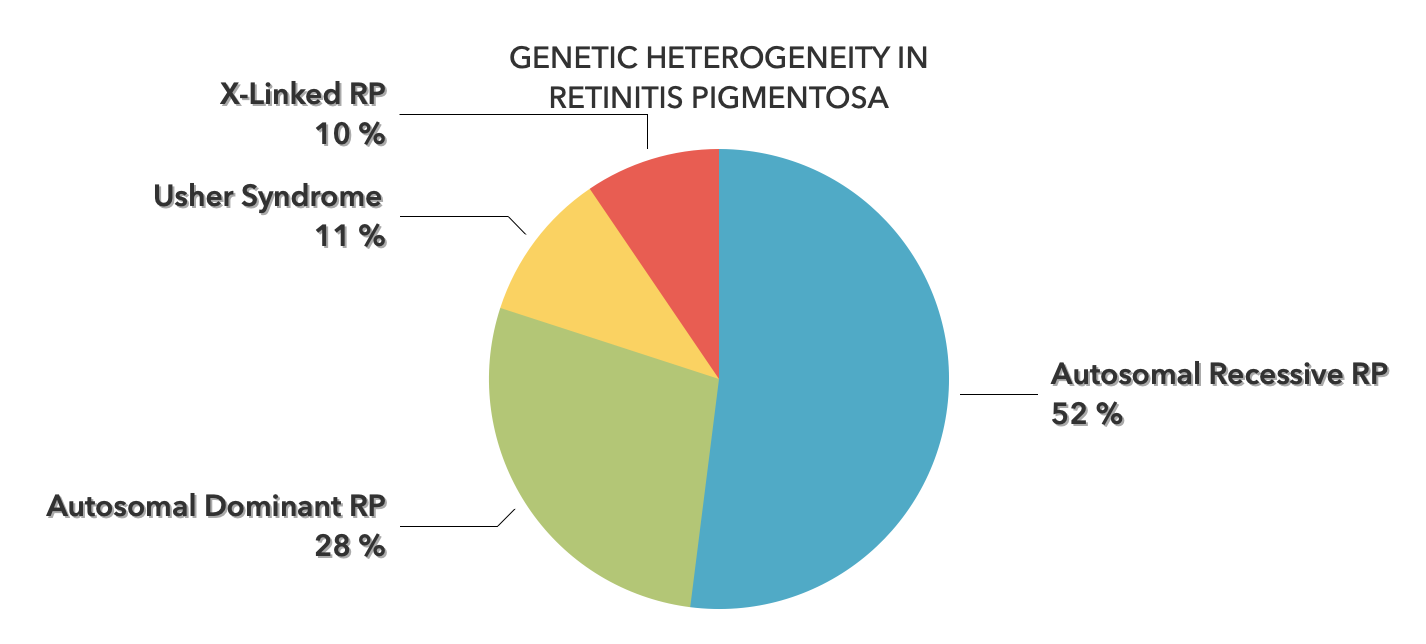
An autosomal dominant inheritance means that only one copy of an altered gene, from either parent, is needed to cause the condition. Offspring of an affected person are essentially guaranteed to have the same condition. The AD form of RP accounts for about 25-40% of RP cases. Approximately 50-75% of ADRP genetic mutations are detectable, depending on the specific population, with few genes accounting for more than 5% of cases. Mutation of the rhodopsin gene – responsible for the photopigment in rods – accounts for 20-25% of ADRP. While RP always affects the peripheral retina, subtypes of ADRP affect the macula to varying degrees. Macular involvement may range from mild disturbance after ~20 years or severe disturbance within ~10 years. Compared to other inheritance modes, ADRP patients have the best long-term prognosis for maintaining central vision.
An autosomal recessive inheritance means that both parental copies of the gene received by the individual must be mutated for the condition to arise. If only one mutated allele is present, the individual does not have the disorder, but is a carrier. The AR form of RP accounts for about 50-60% of RP cases. ARRP consists of a group of early-onset retinal dystrophies overlapping with leber congenital amaurosis. Only about one-third of ARRP genetic mutations have been identified. Mutations of the RPE65 gene cause roughly 11% of ARRP cases. Subtypes of ARRP include: traditional rod-cone dystrophy, severe early-onset, mild late onset, and even senile forms. ARRP typically has a better prognosis than XLRP, but a worse prognosis than ADRP.
With X-linked RP, genetic mutations associated with RP are located on one of the X-chromosomes. Since males only have one X-chromosome, they are preferentially affected over females because in males one mutated allele is sufficient to cause the disorder while females need two mutated alleles to be affected. The XL form of RP accounts for about 10-15% of RP cases. Two genes are known to cause XLRP: RP2 and RP3. The XLRP subtypes include: traditional rod-cone dystrophy, severe early-onset, and moderate late-onset. Generally, XLRP has the most severe course of progression compared to other inheritance modes.
Usher Syndrome is the most common form of syndromic RP, accounting for approximately 10-17% of RP cases. USH is defined by retinitis pigmentosa occurring in combination with deafness and is responsible for half of people with both blindness and deafness. Deafness occurs due to malfunctioning hair cells of the inner ear. Within the United States, the prevalence is about 1:23,000, but is as high as 1:12,500 in countries like Germany. USH has three clinical subtypes that are generally autosomal recessive and differentiated by the extent and onset of hearing loss. USH 1 and USH 2 are more common than USH 3. USH 1 presents with severe deafness at birth along with vestibular and motor dysfunction. USH 2 has moderate deafness, but normal vestibular and motor function. USH 3 has mild, yet progressive, deafness with vestibular dysfunction occurring about half the time. Deterioration of rods before cones causes vision loss peripherally before continuing centrally. Patients also experience night blindness, photophobia, and difficulty transitioning from different lighting situations. The onset of RP appears to be slightly earlier with USH 1 than USH 2 and USH 3. Most of the genes associated with Usher Syndrome have been identified. Overall, Usher Syndrome has been connected with eleven genetic loci and eight different genes. USH 1 is caused by mutations from seven different genes. USH 2 has three known causative genes. One locus has been associated with USH 3. Interestingly, these gene mutations do not always result in Usher Syndrome and the USH2A and USH3A mutations may cause retinitis pigmentosa without deafness.
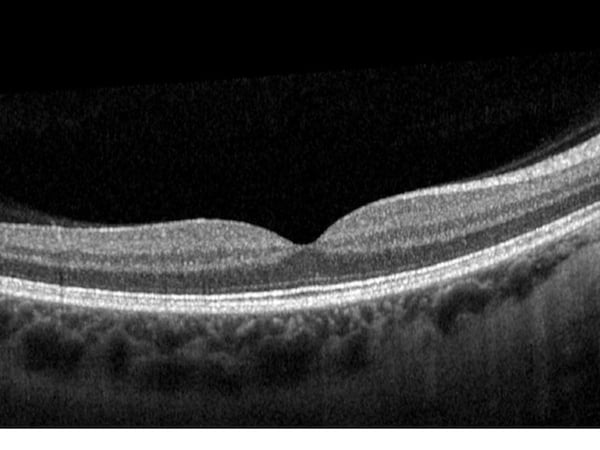
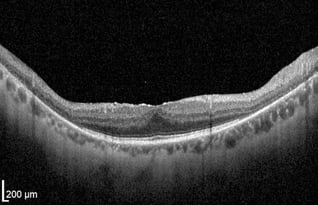
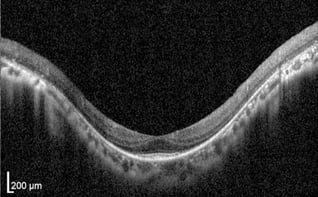
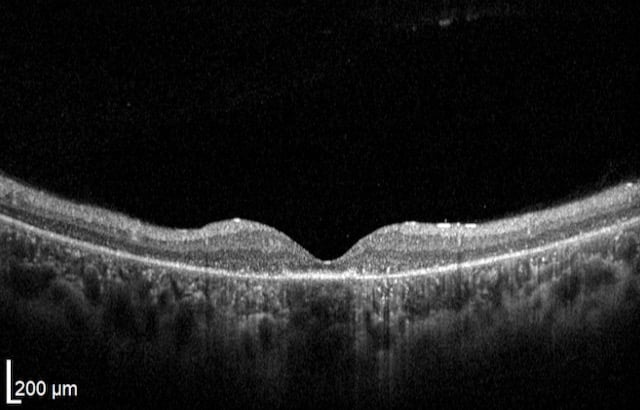
Cone-rod dystrophy is comparable to RP, but opposite. With RP, initial destruction occurs in rods with cones being affected secondarily. In cone-rod dystrophy cones are affected first followed by rods. For this reason, vision loss from cone-rod dystrophy transpires centrally resulting in severe visual impairment earlier. Contributory genes are found in both rods and cones, with greater expression in cones. Common causative genes include: ABCA4, GUCY2D, CRX, RAX2, GNAT2, and PDE6H.
-2.jpeg?width=562&name=Importe%20-%201%20von%201%20(3)-2.jpeg)
The term leber congenital amaurosis (LCA) is often used when RP is identified at birth. LCA is characterized by severe, diffuse rod and cone destruction in the first year of life. Signs include nystagmus, absent pupillary responses, farsightedness, the oculodigital sign, and blindness. Common causative genes include: RPE65, CEP290, GUCY2D, and CRB1. 30% of the time an attributable gene is not found.
%20OCT%20exam%20Restore%20Vision%20Clinic%20.jpg?width=600&name=Congenital%20Stationary%20Night%20Blindness%20(CSNB)%20OCT%20exam%20Restore%20Vision%20Clinic%20.jpg)
CSNB is a congenital, but stable, loss of nighttime vision due to reduced rod photoreceptor and bipolar cell signaling. Other symptoms include nystagmus, strabismus, and high myopia. The majority of cases are X-linked but other causative genes include: GNAT1, SLC24A1, GRM6, TRPM1, CACNA1F, NYX, and LRIT3.
Image: Eye fundus with CSNB in assocciation with severe myopic retinal degeneration
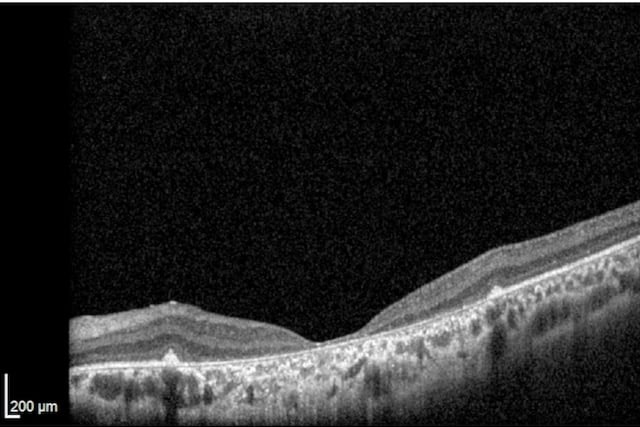
Many macular dystrophies exist including Stargardt Disease, Best Disease, Sorsby Macular Dystrophy, vitelliform degenerations, and pattern dystrophies. Although slowly progressing by nature these conditions account for the majority of central vision loss in persons under the age of 50. Associated genes typically affect cone-rich areas. Causative genes include: PRPH2, PROM1, ELOVL4, ABCA4, BEST1, ARMS2, TIMP3, and EFEMP1.
-2.jpeg?width=586&name=Importe%20-%201%20von%201%20(4)-2.jpeg)
LHON is a mitochondrial inherited retinal disease that presents in childhood or early adulthood in one eye followed by the other eye. Vision loss occurs centrally due to death of retinal ganglion cells and retinal nerve fiber layer atrophy. Causative genes include: ND1, ND4, ND6, and other mitochondrial mutations.
-2.jpeg?width=580&name=Importe%20-%201%20von%201%20(5)-2.jpeg)
ADOA is the most prevalent inherited optic neuropathy. Vision loss presents in early childhood due to retinal ganglion cell death and is typically bilateral (both eyes) and symmetric. These cases are often accompanied by early cataract formation. This condition is autosomal dominant and is primarily associated with the OPA1 and OPA3 genes.

Although the onset of RP is typically during adolescence, the age in which symptoms begin varies substantially. Classical RP presents in adolescence initially as impaired dark adaptation (difficulty adjusting to dim light) before advancing to affect peripheral vision loss beginning in young adulthood. Symptoms frequently go unnoticed by parents with affected children, as kids are quite capable of compensating for their decreased nighttime and peripheral vision early in the disease process. This oftentimes obscures estimations for age of onset. The age of onset is largely dependent on the causative gene(s) in play. RP genes are associated with onsets ranging from less than 5 years of age to greater than 50 years of age. These specific genes are also associated with specific ocular manifestations, such as early maculopathy, bull’s eye maculopathy, dense pigment migration, retinal hypopigmentation, and pericentral pigmentary retinopathy. Age of onset is also linked with other ocular manifestations. For example, nystagmus (shaking of the eyes) is commonly found in early-onset RP patients. Generally early-onset RP progresses more rapidly and is more severe than later presenting forms of RP. The onset and severity is also related to inheritance mode as discussed previously.
Although often difficult for children to articulate, the initial symptom of RP is trouble adjusting to different light levels. Most notably, these patients take much longer to adjust to dim light after being in bright lighting (such as walking into a movie theater on a sunny day). The opposite is true as well, but to a lesser extent (ex. walking out from the movie theater). Diminishing peripheral (usually mid-peripheral) vision is the next symptom experienced. Other less common initial symptoms include photophobia (light sensitivity) and photopsia (flashes of light). Central vision remains unaffected early in the disease course.
The most common and noteworthy symptom of RP is the gradual, yet profound, deterioration of peripheral vision. This occurs throughout the whole periphery, but is concentrated in the mid-periphery and corresponds with the bone-spicule deposits observed in the retina. Peripheral vision is affected before central vision due to RP preferentially targeting rod photoreceptors before cone photoreceptors. Injury of rod photoreceptors also explains the nyctalopia (night blindness) and difficulty with dark adaptation.
-2.jpeg?width=900&name=Importe%20-%201%20von%201%20(6)-2.jpeg)
As RP progresses, vision loss continues encroaching on the central vision and the far peripheral vision. When peripheral vision is severely diminished, yet central vision remains relatively unaffected, this is referred to as tunnel vision. Most forms of RP will eventually lead to tunnel vision, while some progress further and cause central vision deterioration.
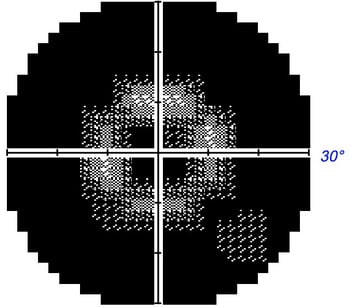 Unlike peripheral vision, central vision loss does not always occur in retinitis pigmentosa. Central vision, if affected at all, remains relatively preserved until later in the disease course, despite the possibility of some abnormal structural or pigmentary changes. Not all genetic forms of RP cause diminished central vision and the severity of central vision deterioration is highly variable. Worsening central vision may begin in middle age due to cone photoreceptor dysfunction in the macula. When central vision is affected, very rarely is all vision completely lost. More than half of RP patients 45 years of age or older are capable of seeing at least 20/40 in one eye. From the same study, only one fourth of RP patients see 20/200 or worse with both eyes, and only 0.5% are completely blind in both eyes.
Unlike peripheral vision, central vision loss does not always occur in retinitis pigmentosa. Central vision, if affected at all, remains relatively preserved until later in the disease course, despite the possibility of some abnormal structural or pigmentary changes. Not all genetic forms of RP cause diminished central vision and the severity of central vision deterioration is highly variable. Worsening central vision may begin in middle age due to cone photoreceptor dysfunction in the macula. When central vision is affected, very rarely is all vision completely lost. More than half of RP patients 45 years of age or older are capable of seeing at least 20/40 in one eye. From the same study, only one fourth of RP patients see 20/200 or worse with both eyes, and only 0.5% are completely blind in both eyes.
One routinely neglected, yet common, symptom in RP is frequent photopsia (flashes of light). Interestingly, this occurs in early and late stages, but less often in moderate stages. Photopsia is experienced by at least 35% of RP patients and is believed to occur due to reduced afferent nerve impulses or spontaneous signaling from inner retina remodeling. Whatever the cause, these flashes of light can be quite frustrating and disturbing for patients.
Photophobia (light sensitivity / glare) is oftentimes more pronounced in advanced RP, as is dyschromatopsia (diminished color vision).
If visual function is severely diminished, to the point of only being able to see hand movements, patients may begin to experience Charles Bonnet Syndrome – famously described as visualizing hallucinations of animate objects or memories.
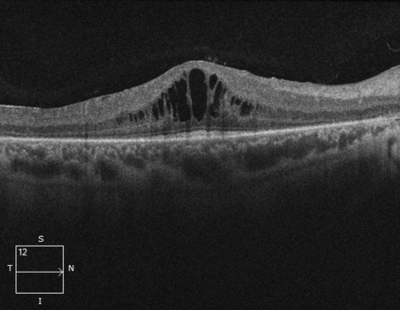 In macular-involving RP, one of the most common complications is cystoid macular edema (CME). Estimated to appear in up to 50% of RP patients, CME occurs when fluid cysts form in the center of the retina and cause retinal swelling or edema. Although painless, CME causes distorted and blurry central vision. Other macular complications with RP include epiretinal membranes (ERM) and macular holes. ERM formation occurs in approximately 36% of RP cases. Macular holes distort and reduce central vision. ERMs may range from benign to causing moderate visual distortion, but may lead to other macular problems. Although non-RP-induced CME is normally only observed in older patients (especially post-operatively), RP-induced CME is found in all age groups. Some studies suggest, but do not confirm, that CME is more prevalent in autosomal dominant RP. Other studies suggest a higher prevalence of CME amongst female RP patients. However the high occurrence of CME in the entire RP population suggests no specific subtype predilection.
In macular-involving RP, one of the most common complications is cystoid macular edema (CME). Estimated to appear in up to 50% of RP patients, CME occurs when fluid cysts form in the center of the retina and cause retinal swelling or edema. Although painless, CME causes distorted and blurry central vision. Other macular complications with RP include epiretinal membranes (ERM) and macular holes. ERM formation occurs in approximately 36% of RP cases. Macular holes distort and reduce central vision. ERMs may range from benign to causing moderate visual distortion, but may lead to other macular problems. Although non-RP-induced CME is normally only observed in older patients (especially post-operatively), RP-induced CME is found in all age groups. Some studies suggest, but do not confirm, that CME is more prevalent in autosomal dominant RP. Other studies suggest a higher prevalence of CME amongst female RP patients. However the high occurrence of CME in the entire RP population suggests no specific subtype predilection.
A diagnosis of retinitis pigmentosa is made through a culmination of patient history, symptoms, ocular findings, and diagnostic testing. Early symptoms begin with difficulty dark adapting and peripheral vision worsening. Nyctalopia (night blindness), photopsia (flashes of light), and photophobia (light sensitivity) may also be present. The classic ocular triad includes bone-spicule pigmentation, retinal vessel attenuation, and waxy optic nerve appearance. Diagnostic testing serves an instrumental role in assessing all hereditary retinal diseases and validating clinical findings. Three main diagnostic instruments utilized for a retinitis pigmentosa diagnosis include perimetry, optical coherence tomography, and electrophysiology.
 Since peripheral eyesight is the predominant form of vision affected with RP, perimetry (aka visual field testing) is essential in management. Visual field loss is usually highly symmetric between the eyes. Perimetry will initially reveal scotomas (missing areas of vision) in the mid-peripheral field of vision. As RP progresses, these scotomas will become more dense and begin spreading both outward (more peripheral) and inward (more central). Peripheral vision worsens at a quicker rate than central vision, which may not be affected at all. Generally at the time of an RP diagnosis, mild to moderate scotomas are present in the mid-periphery and central vision remains unaffected. Perimetry is an indispensable tool for diagnosing and managing RP as with all inherited retinal diseases. Frequent Asked Questions About Visual Field Testing
Since peripheral eyesight is the predominant form of vision affected with RP, perimetry (aka visual field testing) is essential in management. Visual field loss is usually highly symmetric between the eyes. Perimetry will initially reveal scotomas (missing areas of vision) in the mid-peripheral field of vision. As RP progresses, these scotomas will become more dense and begin spreading both outward (more peripheral) and inward (more central). Peripheral vision worsens at a quicker rate than central vision, which may not be affected at all. Generally at the time of an RP diagnosis, mild to moderate scotomas are present in the mid-periphery and central vision remains unaffected. Perimetry is an indispensable tool for diagnosing and managing RP as with all inherited retinal diseases. Frequent Asked Questions About Visual Field Testing
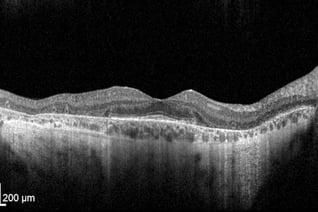
%20(1).png?width=350&name=Alle%20Fotos%20-%201%20von%201%20(2)%20(1).png) As with all retinal and optic nerve conditions, optical coherence tomography (OCT) has become fundamental for RP diagnosis and management. The earliest histopathological changes observable with OCT are shortening of the photoreceptor outer segments and general disorganization of the outer retina.
As with all retinal and optic nerve conditions, optical coherence tomography (OCT) has become fundamental for RP diagnosis and management. The earliest histopathological changes observable with OCT are shortening of the photoreceptor outer segments and general disorganization of the outer retina.
Progression of RP is visualized as continued thinning of the photoreceptor outer segments in addition to diminishing outer nuclear layer thickness. Atrophy of the photoreceptor layer and outer nuclear layer (which contains photoreceptor nuclei) is expected as RP targets rod then cone photoreceptors.
In advanced stages of RP, the photoreceptor outer segments and the outer nuclear layer may wither away completely. While the outer retina exhibits severe disorganization, the inner retina typically remains remarkably intact and may even thicken to some degree.
The bone-spicule pigmentation, from migrating RPE cells, may be demonstrated as hyper-reflected areas in the inner nuclear layer, outer nuclear layer, or subretinal space. The degree of hyper-reflected areas in the outer nuclear layer is associated with visual acuity. Thinning of the photoreceptor outer segments in the macular area is correlated with a decrease in visual field sensitivity. OCT is therefore useful in diagnosing, monitoring, and visualizing structural changes in RP patients.
Furthermore, OCT is valuable in diagnosing other RP-associated macular conditions such as cystoid macular edema (CME), epiretinal membranes (ERM), and macular holes. CME is one of the more common RP-associated retinal findings and can be visualized with OCT as cystic-like fluid spaces in the inner nuclear layer, outer nuclear layer, outer plexiform layer, and ganglion cell layer of the retina.
Read more how to read and understand OCT scans.
 Modern electrophysiology aids in the diagnosis and management of RP by providing quantitative and objective measurements of retinal function. The most useful electrophysiological test is the electroretinogram (ERG) capable of detecting even the slightest irregularities in photoreceptor signaling, oftentimes preceding patient symptomology. Pertinent to RP is the a-wave of the ERG test, which is a measurement of rod and cone electrical signaling captured by flashes of light in a light or dark-adapted environment. Rods are isolated through dark-adaptation while cones are isolated through light-adaptation. In an RP patient, the a-wave is delayed, reduced, or even absent depending on the stage of the disease. Rod photoreceptor function deteriorates earlier than cone photoreceptor function demonstrated by a weaker a-wave in low light than bright light settings. Full-field ERG testing typically demonstrates a 9-11% overall annual rate of decline amongst RP patients with a slower central cone decline of 4-7%. ERG testing is particularly useful for earlier diagnosis and anticipating further progression as changes in electrophysiology precede visual field changes.
Modern electrophysiology aids in the diagnosis and management of RP by providing quantitative and objective measurements of retinal function. The most useful electrophysiological test is the electroretinogram (ERG) capable of detecting even the slightest irregularities in photoreceptor signaling, oftentimes preceding patient symptomology. Pertinent to RP is the a-wave of the ERG test, which is a measurement of rod and cone electrical signaling captured by flashes of light in a light or dark-adapted environment. Rods are isolated through dark-adaptation while cones are isolated through light-adaptation. In an RP patient, the a-wave is delayed, reduced, or even absent depending on the stage of the disease. Rod photoreceptor function deteriorates earlier than cone photoreceptor function demonstrated by a weaker a-wave in low light than bright light settings. Full-field ERG testing typically demonstrates a 9-11% overall annual rate of decline amongst RP patients with a slower central cone decline of 4-7%. ERG testing is particularly useful for earlier diagnosis and anticipating further progression as changes in electrophysiology precede visual field changes.
The differential diagnosis list for RP includes many hereditary retinal diseases. Disorders impairing central vision more than peripheral vision may be ruled out quickly. Other childhood retinal dystrophies may be ruled out based on patient history, clinical findings, diagnostic, and genetic testing. Differentials for RP include:
Additionally, RP is oftentimes a component of a more specific diagnosis (syndromic RP), such as Usher syndrome. Specific types of syndromic retinitis pigmentosa include:
Proper management of retinitis pigmentosa includes not only monitoring and treating the eye, but also providing emotional and psychological support to patients and their families. This may entail longer and empathy-filled patient visits, referrals to psychologists, and support for family members. Low vision referrals are warranted, especially in later disease stages. Genetic testing and counseling is also advisable as family members may be unknowingly affected. Multidisciplinary approaches combining ophthalmological and genetic therapies are helpful. Eye examinations should be performed at least annually, and possibly more frequently depending on the recommendations of your eye care provider.
Management of retinitis pigmentosa is incomplete without proper genetic testing to determine the involved genetic mutations. Genetic testing better informs clinicians of the expected prognosis and potential treatment modalities. Approximately 84 genes are linked with non-syndromic RP. The majority of these genes encode proteins responsible for pathways within the photoreceptor layer and RPE. These proteins play roles in processes such as phototransduction, the visual cycle, or metabolic support of photoreceptors. Since these pathways are delicately and precisely regulated, mutations in these genes disrupt specific retinal processes and the entire visual system.
Identifying the specific pathways involved provides better fundamental understanding of the disease pathogenesis and allows for treatments targeting these precise pathway and protein deficiencies. In recent years, monumental advances have been made identifying causative genetic mutations resulting in RP, largely due to whole exome sequencing. Whole exome sequencing analyzes protein coding and is capable of determining an exact molecular diagnosis in 60-80% of RP cases while whole genome sequencing will further enhance exact molecular diagnosis.
The ideal age for molecular diagnostic testing is debatable, especially in pre-symptomatic cases. Patients and families should understand the benefits and risks of genetic testing that include – but are not limited to – finding a diagnosis with no cure early in life, finding unrelated genetic mutations, or finding a genetic result of unclear significance.
In RP cases, genotyping improves both genetic and ophthalmological counseling and family education. An exact molecular diagnosis may also be useful in predicting the likelihood of central vision deterioration, functional prognosis, and the likelihood of family member involvement.
Treatment options for RP can be divided into two main categories: general therapies targeting the overall condition, and treatments for associated ocular complications. There is currently no cure for retinitis pigmentosa nor is there any way to completely prevent progression, although slowing progression may be possible.
Commonly Used Therapy:
 The most common treatment intervention historically has been Vitamin A supplementation. Vitamin A is essential for photoreceptor function and is utilized poorly in RP. Some studies suggest Vitamin A supplementation results in slower decline in vision loss and slower ERG a-wave reduction (representing photoreceptor function). Although the results are mixed, several studies indicate daily supplementation with 15,000 IUs of Vitamin A may slow vision deterioration in adults with classic RP.
The most common treatment intervention historically has been Vitamin A supplementation. Vitamin A is essential for photoreceptor function and is utilized poorly in RP. Some studies suggest Vitamin A supplementation results in slower decline in vision loss and slower ERG a-wave reduction (representing photoreceptor function). Although the results are mixed, several studies indicate daily supplementation with 15,000 IUs of Vitamin A may slow vision deterioration in adults with classic RP.
Additional studies also indicate that a diet high in omega-3 fatty acids and lutein supplementation combined with Vitamin A supplementation may result in slower visual decline. No study has demonstrated a suppressed rate of visual deterioration with omega-3 fatty acids (usually 200mg) or lutein (usually 12mg) alone, but in combination with Vitamin A supplementation these supplements may be helpful.
Vitamin A supplementation may be beneficial in both early-onset childhood RP and classic RP in adults. Furthermore, diet-based treatment has shown effectiveness in specific syndromic RP types such as adult Refsum disease, Bassen-Kornzweig syndrome, and alpha-tocopherol transfer protein deficiency. Relatedly, oral treatments of certain Vitamin A active metabolites, such as 9-cis-retinoid, are showing promise for maintaining retinal integrity and function.
 Before beginning any supplementation, including Vitamin A, several items must be considered:
Before beginning any supplementation, including Vitamin A, several items must be considered:
One common and easily treatable condition associated with RP is posterior subcapsular cataracts. As with all cataracts, treatment involves surgical intervention in the form of cataract surgery. Visual improvement after this surgery is dependent on macular involvement of the RP and the density of the cataracts. Another common ocular complication of RP is cystoid macular edema (CME). CME is generally treated with topical or oral carbonic anhydrase inhibitors. Topical steroids and NSAIDs may also be utilized. Persistent CME may be treated with intravitreal steroid injections. Epiretinal membranes are generally left untreated unless significantly affecting vision, in which case surgical intervention is used to peel the membrane. Macular holes formation may also be treated surgically.
Visual rehabilitation entails maximizing a patient’s functional vision to enhance their ability to perform activities of daily living. This rehabilitation is primarily performed by low-vision specialists or optometrists and includes orientation and mobility training and use of specialized low-vision devices (handheld magnifiers, electronic magnifiers, telescopes, reverse telescopes, etc) to optimize residual vision. As technology advances, new low vision tools have been developed to assist RP patients. Some examples of this technology include, voice-to-text recognition, eSight and OrCam electronic eyewear, seeing AI (reads books/articles/texts/emails out loud), and applications like WayAround (uses your camera to scan objects/items and identify them out loud) and BeMyEyes (volunteers identify objects for you via your camera phone). These tools are already providing a higher quality of life for many RP patients.
 Note: The information given in this blog are the opinions of the authors and for reader familiarization purposes only. This blog is not intended as a substitute for professional medical advice. Also, the information provided does not replace or abolish any official or legal terms for glaucoma diagnosis, treatment, and management. Authors are not liable for any undesirable consequences or effects related to the information provided in the blog.
Note: The information given in this blog are the opinions of the authors and for reader familiarization purposes only. This blog is not intended as a substitute for professional medical advice. Also, the information provided does not replace or abolish any official or legal terms for glaucoma diagnosis, treatment, and management. Authors are not liable for any undesirable consequences or effects related to the information provided in the blog.
This page prepared in cooperation with Kaleb Abbott, O.D., M.S.
