At Restore Vision Clinic, we understand the profound impact that Leber Congenital Amaurosis (LCA) has on both children and their families. While traditional treatments have been limited, recent advancements offer renewed hope. Notably, pioneering gene therapy trials have demonstrated significant improvements in vision for children with LCA, marking a paradigm shift in treatment approaches.

Complementing these developments, our exclusive Fedorov Restoration Therapy provides a non-surgical, non-invasive approach designed to enhance visual function. By employing gentle electrical stimulation, this therapy aims to optimize the performance of existing visual pathways, potentially improving vision in children affected by LCA.
We are committed to offering personalized care tailored to each child’s unique condition. Our interdisciplinary approach combines insights from ophthalmology and neurology to address complex visual impairments comprehensively.
If your child has been diagnosed with LCA, we encourage you to reach out to us. Our team is ready to discuss how Fedorov Restoration Therapy may offer a path toward improved vision and a better quality of life. Please contact us to schedule a consultation and take the first step toward restoring your child’s sight.
Leber Congenital Amaurosis. An overview.
INTRODUCTION
Not to be confused with Leber Hereditary Optic Neuropathy (LHON), Leber Congenital Amaurosis (LCA) is a rare collection of congenital retinal dystrophies usually presenting within the first 6 months of life. LCA presents with severely reduced central and peripheral vision, sluggish or absent pupillary responses, nystagmus (involuntary eye shaking movements), photophobia (light sensitivity), and hyperopic (farsighted) refractive errors.
Vision deterioration originates from defective development and maintenance of photoreceptors and is linked with at least 25 genetic mutation variants. Leber refers to the German ophthalmologist Theodor Leber who discovered the condition in 1869 while amaurosis refers to unspecified vision loss in the absence of a lesion.
DISEASE AND EPIDEMIOLOGY
Leber Congenital Amaurosis affects approximately 1/40,000 newborns and contributes to 10-18% of congenital blindness cases despite accounting for only 5% of all inherited retinal diseases (IRD). LCA is one of the most severe IRD and typically results in substantial or complete vision loss.

The differentiating characteristic of LCA from other IRD is a near or completely absent photoreceptor response measured with an electroretinogram (ERG). Detailed clinical observation and genetic testing has uncovered a wide spectrum of disease phenotypes and genotypes with varying severity and progression. In addition to nystagmus and light sensitivity, children may also exhibit the “oculodigital sign” consisting of poking, pressing, or rubbing of the eyes using a finger or knuckle. This “oculodigital sign” is claimed to be essentially pathognomonic for LCA. Though vision is generally stable or slowly progressing, one third of children with LCA demonstrate no light perception and the majority have best corrected vision of 20/400 or worse. Onset, rate, and degree of vision loss vary markedly depending on the involved genetic variants. LCA generally occurs in isolation without systemic involvement, however the significantly reduced vision may contribute to certain developmental delays.
ETIOLOGY
LCA is a group of hereditary retinal diseases usually occurring in autosomal recessive fashion, but may be autosomal dominant as well. 17 loci and 25 genes have been linked with LCA.
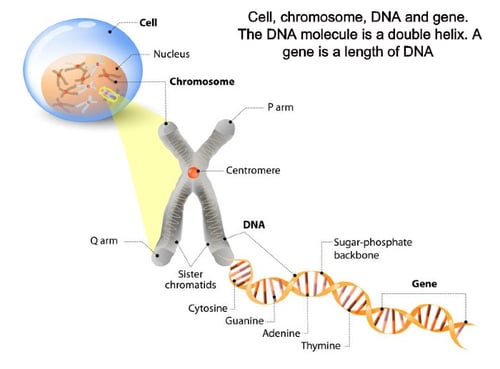
Genes associated with LCA (with known estimated frequency) include: GUCY2D (10-20%), RPE65 (5-10%), SPATA7 (3%), AIPL1 (<5%), LCA5 (1-2%), RPGRIP1 (5%), CRX (1%), CRB1 (10%), NMNAT1, CEP290 (15-20%), IMPDH1 (5%), RD3 (<1%), RDH12 (10%), LRAT (<1%), TULP1 (<1%), KCNJ13, GDF6, OTX2, CABP4, CLUAP1, IQCB1, DTHD1, IFT140, ALMS1, and PRPH2. These genes are involved with retinal and physiological pathways, specifically photoreceptor development and visual transduction.
PATHOPHYSIOLOGY
Vision deterioration in LCA arises from inability of the visual cycle to convert photons into visual signals largely due to photoreceptor dysfunction. Photoreceptors are responsible for phototransduction of light into electrical signals that are then propagated to the visual cortex for image formation. Genes associated with LCA are almost exclusively involved in the retina or the retinal pigment epithelium and are responsible for functions such as photoreceptor morphogenesis, photoreceptor maintenance, phototransduction, protein transport, outer segment phagocytosis, cell-to-cell interactions, and ciliary transport. Genetic mutations involved in these processes result in photoreceptor maldevelopment, degeneration, dysfunction, and death.
RISK FACTORS
Risk factors include having family members with LCA, specifically both parents having a carrier mutation or a one parent having an autosomal dominant mutation.
GENETICS
 Recent molecular genetic developments have led to successful mapping and identification of 17 loci and 25 distinct genes linked with LCA. These genes and their protein products are responsible for photoreceptor development and visual transduction.
Recent molecular genetic developments have led to successful mapping and identification of 17 loci and 25 distinct genes linked with LCA. These genes and their protein products are responsible for photoreceptor development and visual transduction.
In typical autosomal recessive nature, LCA inheritance arises from each parent passing on a single copy of the mutant recessive allele. When both parents are unaffected carriers, each offspring has a 25% risk of developing LCA. Although most known genetic defects associated with LCA are autosomal recessive, a few exhibit autosomal dominant patterns. In which case, only one genetic mutation needs to be present for LCA to occur.
Pathological gene variants have been identified in 70-80% of LCA cases, but genetic heterogeneity continues to be an obstacle in identifying the remaining variants. While disease trajectory and phenotypic expressions vary between genotypes, considerable phenotypic overlap exists between these genotypes, further complicating identification of the remaining pathological mutations. Mutation frequencies also vary between different ethnic groups. Genetic testing is used to pinpoint which pathological variant is involved for individuals and family members. Result possibilities for genetic testing are detailed below.
Genetic testing results possibilities:
- Positive: A pathological variant known to cause a given genetic disorder has been identified. Molecular diagnosis is attainable, familial risk can be evaluated, and preventative or therapeutic measures can be initiated.
- Inconclusive: A genetic variant of unknown significance was detected. Familial genetic testing is warranted and further examination is advisable.
- Negative: A genetic variant was not found. This finding does not negate a clinical diagnosis and it is possible that other unidentifiable genetic alterations or variations may still be at play.
- Unexpected: Unanticipated results were discovered such as consanguinity, no family correlation, or other genetically based diseases.
Genetic counseling: Due to LCA being predominantly autosomal recessive, family members of those with LCA may not be aware that they are carriers. Carriers (heterozygotes) usually remain asymptomatic with no retinal findings, but may demonstrate mild reduction of ERG responses.  Genetic counseling provides families with information regarding the inheritance, implications, and the condition itself. In addition to encouraging genetic testing of family members, this information empowers families to make educated decisions regarding medical and personal decisions. Unaffected parents are heterozygotes carrying one mutant allele. Their offspring have a 25% chance of being affected, 50% chance of being a carrier, and a 25% chance of being unaffected. Unaffected siblings have a 2/3 risk of being a carrier. Siblings of unaffected parents also have a 50% chance of being a carrier. Offspring of those with LCA will, at the very least, be carriers. Therefore, carrier testing is warranted for family members. Familial risks are more pronounced if the LCA gene is autosomal dominant. Moreover, genetic counseling is recommended as certain subtypes of LCA may genetically predispose patients to additional systemic conditions.
Genetic counseling provides families with information regarding the inheritance, implications, and the condition itself. In addition to encouraging genetic testing of family members, this information empowers families to make educated decisions regarding medical and personal decisions. Unaffected parents are heterozygotes carrying one mutant allele. Their offspring have a 25% chance of being affected, 50% chance of being a carrier, and a 25% chance of being unaffected. Unaffected siblings have a 2/3 risk of being a carrier. Siblings of unaffected parents also have a 50% chance of being a carrier. Offspring of those with LCA will, at the very least, be carriers. Therefore, carrier testing is warranted for family members. Familial risks are more pronounced if the LCA gene is autosomal dominant. Moreover, genetic counseling is recommended as certain subtypes of LCA may genetically predispose patients to additional systemic conditions.
DIAGNOSIS
Diagnosis of LCA is ordinarily made by an ophthalmologist based on history, clinical findings, and diagnostic procedures. Clinical testing includes estimation of central visual acuity, peripheral awareness, pupil responses, refractive error status, and dilated fundus examination. An electroretinogram (ERG) is utilized to demonstrate reduced photoreceptor response while optical coherence tomography (OCT) provides cross-sectional scans and thickness measurements of the retinal layers.
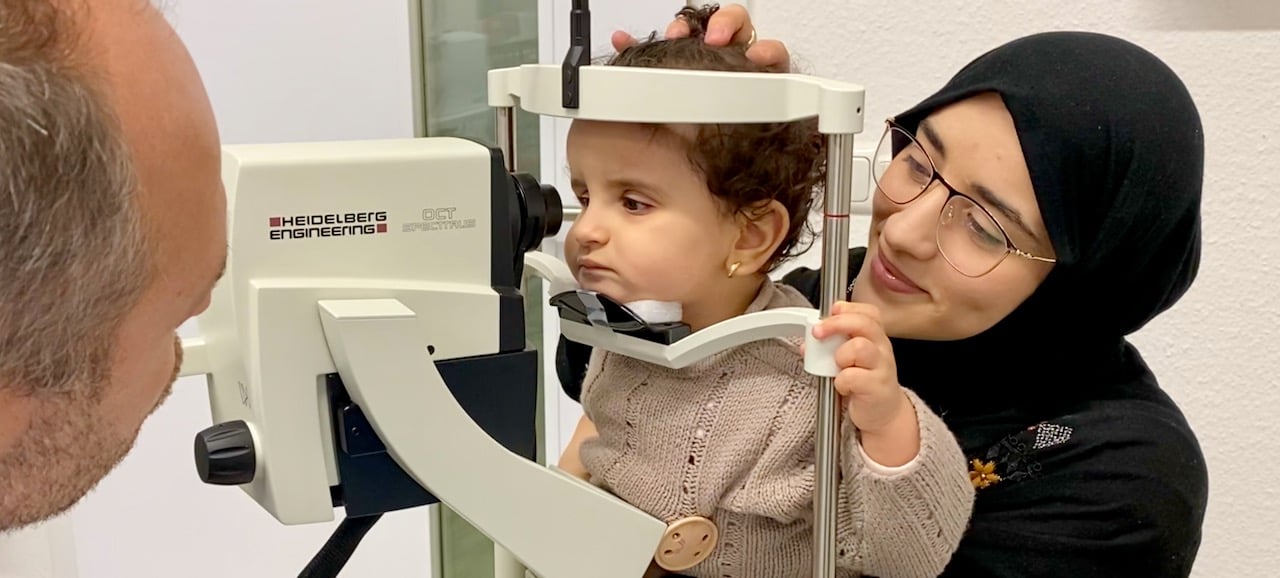
Genetic testing, performed approximately 80% of the time, is warranted in cases of suspected LCA and proves useful in diagnosis, prognosis, treatment options, and family planning.
CLINICAL SIGNS
Apart from perceptible visual impairment, the most prevalent early symptom of LCA is nystagmus present in all positions of gaze. Extreme photophobia and deficient eye tracking frequently manifest within the first year or life. Observers might also notice the oculodigital sign. Eye care providers may detect sluggish or absent pupillary responses to light. Retinoscopy or auto-refraction will likely reveal an abnormally high degree of hyperopia (>+5.00 diopters), likely from impaired emmetropization.
Children will exhibit severe visual impairment and light aversion within the first 6 months of life.
RETINAL FINDINGS. LCA features a deceptively benign fundus appearance in early stages, which may not be particularly useful for initial diagnosis. Even after progression, fundus appearance remains variable.
-2.jpeg?width=562&name=Importe%20-%201%20von%201%20(3)-2.jpeg)
As with many IRD, increased retinal pigmentation and vessel attenuation are common retinal findings. In later stages, optic disc pallor, chorioretinal atrophy, pigmentary retinopathy, maculopathy, and retinal pigment epithelial changes may be observed. Ophthalmoscopy may also reveal fundus hypopigmentation, a “marbled” fundus, optic disc edema, disc drusen, “bone-spicule” pigment migration, exudates, or retinal telangiectasia. Retinal findings differ by case due to numerous causative genotypes with differing phenotypic characteristics.
DIAGNOSTIC PROCEDURES. Electrophysiologic Testing is a gold standard for kids or infants who are unable to verbalize visual complaints testing unveils severely subnormal or undetectable outer retinal (photoreceptor) responses. This severe reduction or absence of a photoreceptor response within the first year of life is unique to LCA and differentiates LCA from other IRD. OCT autofluorescence may display lipofuscin accumulation from undegradable photoreceptor byproduct accumulation while cross sectional scans may demonstrate abnormal retinal architecture. Visual field testing is not obtainable with these young patients.
LABORATORY TESTS. Modern day LCA diagnosis involves not only clinical evaluation and diagnostic procedures, but also genetic testing. To date, 25 genes are known to account for 70-80% of LCA cases. Certain genetic mutations have specific phenotypic characteristics such as a blonde fundus, peripheral white punctate lesions, dense intraretinal pigmentation, or macular atrophy. Genetic testing informs a more exact diagnosis and prognosis, thus aiding in proper treatment modality selection and family education.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis includes a wide range of other IRD and certain systemic conditions (especially neurological, nephrological, hepatological, metabolic, and peroxisomal conditions).
Such differentials include:
- Early onset retinal dystrophy
- Cone-rod dystrophy
- Pigmentary retinopathy
- Achromatopsia
- S-cone monochromatism
- Disorders of mitochondrial dysfunction
- Optic atrophy
- Ocular albinism
- Congenital stationary night blindness
- Early onset retinitis pigmentosa
- Diabetes mellitus
- Cerebellar and corpus callosum hypoplasia
- Joubert Syndrome
- Cororenal syndrome
- Senor-Loken Syndrome
- Zellweger Syndrome
- Bardet–Biedl syndrome
- Neonatal adrenoleukodystrophy
- Infantile Refsum disease
- Abetalipoproteinemia
- Hyperthreoninemia
VISUAL IMPAIRMENT
LCA produces substantial vision deterioration from infancy. One third of children with LCA demonstrate no light perception and the majority will have vision of 20/400 or worse. The rate and degree of vision loss varies markedly depending on the genotypes involved. Studies indicate that the later the onset of visual symptoms, the greater the likelihood of retaining some vision.
COMPLICATIONS
Although the exact mechanisms are unknown, certain ocular complications are known to exist with LCA including keratoconus, cataracts, strabismus, and glaucoma. Recession of the eye into the orbit, or enophthalmos, may also occur from the excessive eye rubbing, pressing, and poking. Such behaviors may also increase the likelihood of keratoconus formation as this corneal degeneration has been shown to develop and progress from aggressive eye rubbing. Sadly, despite the debilitating nature of these associated ocular conditions, rarely do these complications become visually limiting due to the already severely reduced vision from LCA.
Intellectual disability has long been reportedly associated with LCA. The connection between LCA and mental deficiency, neurodevelopmental delays, and oculomotor apraxia behaviors remains questionable. While LCA usually occurs in isolation, approximately 20% of patients with LCA proceed to develop mental retardation. It is plausible, however, that the severely reduced vision from LCA contributes to delayed or hindered developmental skills, as vision is a key component in learning and developing.
MANAGEMENT
Due to the complexity of IRD and sheer overwhelming number of subtypes and associated genes, eye care practitioners are prone to mismanaging these patients. Patients are oftentimes diagnosed with a retinal dystrophy; told it leads to progressive vision loss or blindness; informed that no viable treatments exist; and told it is likely genetic so family members may be affected too. These patients and families are left feeling hopeless and neglected. Traditionally, if any treatment was initiated, such intervention aimed to slow progression through use of nutritional supplements or maximize remaining vision through low-vision devices.
Medical Follow-up: With advances in molecular testing and gene replacement therapy, IRD specialists and centers are emerging which are capable of providing accurate, genetically based diagnosis and prognosis. Such referrals are merited and benefit not only patients but also future research. Regular comprehensive eye examinations should monitor for any visual or retinal changes. Refractive errors should be ascertained and correspondingly treated. Low vision specialists ought to be involved so remaining vision may be optimized. Secondary complications may be mitigated through parent education, especially from eye rubbing or poking. Early involvement with a developmental pediatrician and therapy services is indicated to promote developmental skills. Appropriate school and workplace modifications may optimize success for these patients.
PROGNOSIS
Generally, prognosis for LCA patients is exceedingly poor. Although few patients retain ambulatory vision, the majority of patients lose functional vision early in childhood. Vision testing rarely demonstrates improvement over time.
Even LCA is incurable medical condition, there is a progress in field of partial vision restoration based on medical approach aimed to activate functional state of partially damaged but preserved photoreceptors - electrical stimulation therapy which a core part of Fedorov Restoration Therapy. To read more click here.
CONCLUSIONS
Leber Congenital Amaurosis is a group of congenital retinal dystrophies similar to other inherited retinal diseases but with earlier, and oftentimes, more pronounced vision deterioration. LCA is an inherited disease linked with 25 known genetic mutations normally occurring in an autosomal recessive fashion. Although historically the prognosis of patients with LCA and other inherited retinal diseases was exceedingly poor, the advent of specific molecular diagnosis and novel treatment modalities provides hope for not only delaying vision deterioration, but also reversing vision loss. Furthermore, alternative treatment models are emerging such as photoreceptor transplantation and electro-stimulation therapy, demonstrating potential for restoring previously lost vision in these patients.
Blog prepared in cooperation with Kaleb Abbott, O.D., M.S.
REFERENCES
- Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147-1154.
- Cideciyan AV, Jacobson SG. Leber Congenital Amaurosis (LCA): Potential for Improvement of Vision. Invest Ophthalmol Vis Sci. 2019;60(5):1680-1695.
- Artur V. Cideciyan, Samuel G. Jacobson; Leber Congenital Amaurosis (LCA): Potential for Improvement of Vision. Invest. Ophthalmol. Vis. Sci.2019;60(5):1680-1695. doi: https://doi.org/10.1167/iovs.19-26672.
- Abeshi, A., Coppola, P., Beccari, T., Dundar, M., Falsini, B., & Bertelli, M. (2017). Genetic testing for Leber congenital amaurosis, The EuroBiotech Journal, 1(s1), 63-65. doi: https://doi.org/10.24190/ISSN2564-615X/2017/S1.20
- Weleber RG, Francis PJ, Trzupek KM, et al. Leber Congenital Amaurosis. 2004 Jul 7 [Updated 2013 May 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1298/
- Hufnagel RB, Ahmed ZM, Corrêa ZM, Sisk RA. Gene therapy for Leber congenital amaurosis: advances and future directions. Graefes Arch Clin Exp Ophthalmol. 2012;250(8):1117-28.
- Leber congenital amaurosis. Genetics Home Reference. August 2010. Retrieved 14 May 2017.



2 Comments
Click here to read/write comments